
Abstract
Alteration of endoplasmic reticulum (ER) Ca2+ homeostasis leads to excessive cytosolic Ca2+ accumulation and delayed neuronal cell death in acute and chronic neurodegenerative disorders. While our recent studies established a protective role for SK channels against excessive intracellular Ca2+ accumulation, their functional role in the ER has not been elucidated yet. We show here that SK2 channels are present in ER membranes of neuronal HT-22 cells, and that positive pharmacological modulation of SK2 channels with CyPPA protects against cell death induced by the ER stressors brefeldin A and tunicamycin. Calcium imaging of HT-22 neurons revealed that elevated cytosolic Ca2+ levels and decreased ER Ca2+ load during sustained ER stress could be largely prevented by SK2 channel activation. Interestingly, SK2 channel activation reduced the amount of the unfolded protein response transcription factor ATF4, but further enhanced the induction of CHOP. Using siRNA approaches we confirmed a detrimental role for ATF4 in ER stress, whereas CHOP regulation was dispensable for both, brefeldin A toxicity and CyPPA-mediated protection. Cell death induced by blocking Ca2+ influx into the ER with the SERCA inhibitor thapsigargin was not prevented by CyPPA. Blocking the K+ efflux via K+/H+ exchangers with quinine inhibited CyPPA-mediated neuroprotection, suggesting an essential role of proton uptake and K+ release in the SK channel-mediated neuroprotection. Our data demonstrate that ER SK2 channel activation preserves ER Ca2+ uptake and retention which determines cell survival in conditions where sustained ER stress contributes to progressive neuronal death.
Similar content being viewed by others

Main
In eukaryotic cells, the endoplasmic reticulum (ER) is the subcellular site of protein folding and maturation1 and the main intracellular calcium store of the cell. Since ER resident chaperones required for protein folding need high calcium concentrations for their activity, alteration of the ER calcium ([Ca2+]ER) homeostasis results in an imbalance between the capacity of the protein processing machinery and the amount of ER accumulating unfolded proteins, thereby leading to ‘ER stress’.2, 3 The increasing number of unfolded proteins inside the ER lumen provokes the dissociation of 78 kDa glucose-regulated protein (grp78) from three ER transmembrane receptors, namely PRKR-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1), thereby initiating the unfolded protein response (UPR). This ER-specific stress response aims to maintain cell survival through different molecular pathways, such as attenuation of the translation initiation rate, enhanced protein folding or elimination of misfolded proteins.4 Under conditions of prolonged or severe ER stress, however, the UPR switches from homeostatic feedback regulation towards proapoptotic signaling.5 UPR was detected in post-mortem human brains of so-called protein-misfolding disorders, such as Parkinson’s and Alzheimer’s disease, suggesting that prolonged ER stress contributes to neurodegeneration.6 Different pharmacological compounds modulated the UPR and protected against ER stress-induced apoptosis.7 These include antioxidants8, 9 indicating a close relation between ER stress and oxidative damage.10 Additionally, pharmacological applications that interfere with alterations of the intracellular calcium homeostasis have a protective potential against ER stress-induced cell death.11, 12
Several ER-located potassium channels are supposed to be important for [Ca2+]ER uptake and release by maintenance of the countercurrent required for electroneutrality.13 For instance, small-conductance calcium-activated potassium (SK) channels have recently been detected in the sarco-/endoplasmic reticulum of cardiomyocytes and neurons, respectively, where they may regulate [Ca2+]ER uptake.14 However, the role of SK channels in conditions of [Ca2+]ER disturbances and stress has not been elucidated yet. In the present study, we aimed to demonstrate the impact of SK2 channel activation on ER stress responses and the respective cell death pathways.
Results
Positive modulation of SK2 channels protects against brefeldin A-induced apoptosis in HT-22 cells
To study the role of SK channel activation on ER stress-mediated apoptosis, we first established a model for ER stress in immortalized HT-22 mouse hippocampal neurons.15 To promote ER stress in HT-22 cells, we applied brefeldin A, a pharmacological ER stress inducer, which inhibits the protein trafficking in the endomembrane system and leads to accumulation of unfolded proteins in the ER lumen.16 We challenged HT-22 cells for 24 h with brefeldin A at concentrations ranging from 1.5 to 5 μM (Figure 1a) and determined pronounced changes in cell morphology, cell viability as well as apoptotic cell death with 5 μM brefeldin A (Figure 1a–d). To investigate the effects of SK channel activation in this model of ER stress-induced cell death, we applied cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine (CyPPA), a positive modulator of SK channels, selective for SK2 and SK3.17 Since SK3 channels are not expressed in immortalized HT-22 cells,18 the use of CyPPA allowed selective investigation of SK2 channel activation. Co-application of CyPPA (50 μM) with brefeldin A mediated a slight protection against ER stress-induced cell death (Supplementary Figure S1A). However, pretreatment with CyPPA for 24 h inhibited the brefeldin A-induced changes in cell morphology (Figure 1b) and significantly reduced brefeldin A-mediated cell death at CyPPA concentrations ranging from 12.5 to 50 μM (Supplementary Figure S1B), with 50 μM being the most potent concentration (Figure 1c and d). We also investigated the effects of SK channel activation in other models of ER stress-induced toxicity, such as tunicamycin and thapsigargin. CyPPA protected cells against tunicamycin treatment (Supplementary Figure S2A), an inhibitor of N-linked glycosylation, but not against the irreversible sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) blocker thapsigargin (Supplementary Figure S2B). To exclude substance-specific effects of CyPPA we applied two additional activators of SK channels, namely NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) and 1-EBIO (1-ethylbenzimidazolinone), and found significant reduction in brefeldin A and tunicamycin-induced toxicity, similar to the findings with CyPPA (Supplementary Figure S3).

Pharmacological activation of SK2 channels with CyPPA protects hippocampal HT-22 neurons against ER stress-induced apoptosis. (a) Cell viability of HT-22 cells challenged with different concentrations of brefeldin A measured by MTT assay (mean±SD, n=8; **P<0.01, ***P<0.001 compared with untreated control cells). (b–d) HT-22 cells were treated with brefeldin A (5 μM) with and without CyPPA pretreatment (50 μM, 24 h). (b) Representative pictures of HT-22 cell morphology after 24 h treatment. (c) Representative pictures of flow cytometric analysis after AnnexinV-FITC/PI double staining. Cells in the lower right quarter indicate AnnexinV-positive, early apoptotic cells. Cells in the upper right quarter indicate AnnexinV-positive/PI-positive, late apoptotic cells. (d) Quantification of cell viability after 24 h treatment (mean±S.D., n=3; ***P<0.001 compared with untreated control cells, ###P<0.001 compared with brefeldin A-only treated cells)
Next, we analyzed the effects of brefeldin A and CyPPA on caspase-12 cleavage. In murine cells, pro-caspase-12 is supposed to be mainly located in the ER membrane and being specifically cleaved under conditions of ER stress by calpains owing to increasing cytoplasmic calcium ([Ca2+]cyto) concentrations.19, 20 Active caspase-12 activates the caspase cascade and ultimately induces apoptosis in a mitochondrial cytochrome c-independent manner.21 In our studies, CyPPA alone did not affect the basal [Ca2+]cyto levels, but reduced the moderate increase in [Ca2+]cyto detected in brefeldin A-stimulated HT-22 cells (Figure 2a). Furthermore, treatment with brefeldin A activated caspase-12 and caspase-3, while CyPPA pretreatment inhibited this effect (Figure 2b–e). The use of the pan-caspase inhibitor Q-VD-OPh revealed the caspase dependency of brefeldin A-mediated cell death (Figure 2f and g). According to these data, SK2 channel activation reduces ER stress-induced and caspase-mediated cell death in neuronal HT-22 cells.

SK2 channel activation inhibits the caspase cascade. (a) Quantification of Fluo-4 fluorescence after 24 h (mean±S.D., n=3; ***P<0.001 compared with untreated control cells, ###P<0.001 compared with brefeldin A-only treated cells). (b–e) Representative western blot analysis of whole-cell extracts shows brefeldin A-mediated cleavage of caspase-12 and caspase-3 at indicated time points in the presence and absence of CyPPA pretreatment; densitometric analysis of cleaved caspase-3 (24 h) and cleaved caspase-12 (4 h) in relation to the respective loading controls (mean±S.D., n=3). (f and g) Influence of caspase inhibition with Q-VD-OPh on cell viability was detected by AnnexinV/PI double staining and flow cytometric analysis (mean±S.D., n=3; ###P<0.001 compared with brefeldin A-only treated cells)
Activation of SK2 channels regulates the UPR system
Prolonged ER stress stimuli are associated with a sustained activation of the UPR pathway and its switch into proapoptotic signaling.22 Therefore, we investigated next, whether SK2 channel activation by CyPPA may affect the UPR signaling under conditions of ER stress. Our main interest focused on the PERK pathway, which involves induction of the activating transcription factor 4 (ATF4) thereby controlling the expression of genes involved in cell death pathways, including CCAAT/enhancer-binding protein homologous protein (CHOP) and growth arrest and DNA damage-inducible protein GADD34. Additionally, we investigated ER-stress responses through unconventional mRNA splicing of X-box binding protein 1 (XBP-1), a transcription factor for genes involved in protein secretion and folding.23 We found that brefeldin A promoted splicing of XBP-1 mRNA as well as induction of the proteins ATF4, CHOP and GADD34 within 4–6 h of ER stress induction, as measured by reverse transcription polymerase chain reaction (RT-PCR) and western blotting, respectively (Figure 3a–d). The splicing of XBP-1 mRNA was almost completely prevented by CyPPA pretreatment (Figure 3a). Interestingly, CyPPA pretreatment further enhanced the induction of CHOP protein (Figure 3c) but did not affect the CHOP mRNA level (Supplementary Figure S4), while it reduced the amount of ATF4 (Figure 3b) and GADD34 protein (Figure 3d) in HT-22 cell lysates. To address the role of CHOP and ATF4 in brefeldin A-induced cell death, we applied siRNA for specific gene silencing of these ER stress factors (Supplementary Figure S5). In particular, we sought to investigate whether upregulation of the transcription factor CHOP was essential for the brefeldin A-induced apoptosis in HT-22 neurons and, additionally, whether the CyPPA-mediated CHOP expression was involved in the protective effects of the SK channel activator. After siRNA-mediated CHOP knockdown, we only detected a marginal reduction in brefeldin A-induced apoptosis, suggesting that CHOP induction might only play a minor role in brefeldin A toxicity in HT-22 cells. Furthermore, the protective effects of CyPPA were not altered in CHOP-deficient cells (Figure 3e). In contrast to CHOP siRNA, knockdown of ATF4 significantly reduced brefeldin A-induced cell death in HT-22 cells (Figure 3f). Therefore, we concluded a causal role for ATF4 and its downstream target GADD34, but not CHOP, in the detrimental effects of brefeldin A-induced ER stress in HT-22 cells.

SK2 channel activation modulates the UPR of ER-stressed HT-22 cells. Cells were treated with brefeldin A (5 μM) for the indicated time with and without CyPPA pretreatment (50 μM, 24 h). (a) Representative picture and densitometry (mean±S.D.; n=3) of RT-PCR analysis of XBP-1 mRNA (205 bp) and its unconventional splice variant (179 bp). (b–d) Representative western blots and densitrometric analysis of ATF4 (mean±S.D.; n=3), CHOP (n=4) and GADD34 protein (n=3). (e and f) Influence of CHOP siRNA (40 nM), ATF4 siRNA (40 nM) and scrambled (Scr) siRNA (40 nM) on cell viability was detected by AnnexinV/ PI double staining and flow cytometric analysis (mean±S.D.; n=3; n.s.P>0.05, ###P<0.001 compared with brefeldin A-treated cells transfected with scrambled siRNA)
ER stress-associated reactive oxygen species formation is reduced by SK2 channel activation
During ER stress, the increased reactive oxygen species (ROS) production in the ER lumen has been attributed to the enhanced disulfide bond reduction of the accumulating unfolded proteins. In addition, depletion of glutathione through disulfide bond formation stimulates mitochondrial oxidative phosphorylation which leads to enhanced mitochondrial ROS production, thereby further contributing to the impaired cellular redox homeostasis and death.24 Here, brefeldin A strongly increased the percentage of HT-22 neurons with high levels of cytosolic ROS, measured by the fluorescent dye CM-H2DCFDA (Figure 4a and b). To address the question whether brefeldin A was able to promote the production of mitochondrial superoxides ([O2−]m) we challenged the cells with brefeldin A for 24 h and analyzed the fluorescence of MitoSOX, an indicator of [O2−]m. Brefeldin A moderately increased the percentage of HT-22 cells with enhanced [O2−]m production after 24 h of treatment (Figure 4c and d).

Oxidative stress resulting from brefeldin A treatment is reduced by SK2 channel activation. Cells were treated with brefeldin A (5 μM) for 24 h in the presence and absence of CyPPA (50 μM, 24 h). (a and b) Cytosolic ROS was detected by CM-H2DCFDA staining and flow cytometric analysis (mean±S.D.; n=3; ##P<0.01 compared with brefeldin A-only treated cells). (c-d) MitoSOX was used to analyze [O2−]m formation via flow cytometry (mean±S.D.; n=3, *P<0.05 compared with untreated control, #P<0.05 compared with brefeldin A-only treated cells). (e–f) Influence of antioxidative treatment with Trolox (50 μM) on cell viability was detected by AnnexinV/PI double staining and flow cytometric analysis (mean±S.D., n=3; n.s.P>0.05 compared with brefeldin A-only-treated cells)
In contrast, SK2 channel activation attenuated the formation of cytosolic ROS induced by brefeldin A, measured by CM-H2DCFDA fluorescence (Figure 4a and b). Interestingly, we found that CyPPA alone induced a small but significant increase in the percentage of cells with increased [O2−]m (Figure 4c and d). However, CyPPA pretreatment was able to block brefeldin A-mediated [O2−]m formation (Figure 4c and d).
To investigate whether inhibition of ROS formation alone is sufficient to prevent brefeldin A-induced cell death, we applied the antioxidant Trolox and subsequently investigated cell death. Antioxidative treatment could not reduce brefeldin A-mediated cell death (Figure 4e and f), indicating that a reduced ROS production alone is not sufficient for cellular survival in conditions of lethal ER stress.
CyPPA modulates mitochondrial bioenergetics but does not restore brefeldin A-mediated mitochondrial membrane depolarization
In early stages of ER stress, organelle coupling between ER and mitochondria facilitates enhanced mitochondrial respiration and ATP production.25 On the other hand, the physical interaction of ER and mitochondria determines mitochondrial calcium overload and changes in mitochondrial morphology under conditions of prolonged ER stress.26 Thus, we investigated the effects of SK2 channel activation on mitochondrial morphology and bioenergetics in ER-stressed HT-22 cells. It is well established that neuronal cell death is accompanied by fragmentation of the tubular mitochondrial network, loss of mitochondrial membrane potential (ΔΨm) and associated defects in mitochondrial bioenergetics.18, 27 Brefeldin A (24 h) increased the amount of cells showing shortened and fragmented mitochondria in the perinuclear region (Figure 5a and b), indicating mitochondrial damage in dying cells.28 Additionally, brefeldin A exposure resulted in a slight depolarization of ΔΨm, decreased basal mitochondrial respiration, ATP production and spare respiratory capacity (Figure 5c–e). Treatment with CyPPA for 24 h had no impact on the ATP production as measured by luciferase-produced luminescence. However, prolonged treatment with CyPPA for 48 h lowered the generation of ATP (Figure 5d). In line with our previous results,29 CyPPA (24 h) strongly reduced the mitochondrial respiration (Figure 5c). Interestingly, CyPPA-induced loss of mitochondrial respiration was accompanied by an initial glycolytic boost detected by measurements of the extracellular acidification rate as an indicator of glycolysis (Supplementary Figure S6). Although CyPPA pretreatment preserved mitochondrial integrity under conditions of prolonged ER stress (Figure 5a and b), it did not prevent the brefeldin A-induced slight depolarization of ΔΨm (Figure 5e). Moreover, brefeldin A-challenged cells pretreated with CyPPA showed no significant differences regarding mitochondrial respiration and ATP production compared with CyPPA-only treated cells (Figure 5c and d).

CyPPA modulates mitochondrial bioenergetics and function. (a) Representative pictures of mitochondria from HT-22 cells challenged with brefeldin A (5 μM, 24 h) with and without CyPPA pretreatment (50 μM, 24 h). Scale bar 20 μm. (b) Assessment of mitochondrial morphology by dividing into three categories (category I: tubulin-like elongated mitochondria; category II: shortened mitochondria distributed throughout the cytosol; category III: short, fragmented mitochondria surrounding the nucleus). Five hundred cells were evaluated per condition. Values are given from three independent experiments (mean±S.D.; ##P<0.01 compared with brefeldin A-only treated cells). (c) OCR was determined using a Seahorse XF96 Metabolic Flux analyzer. HT-22 cells were treated with brefeldin A (5 μM, 16 h) or/and CyPPA (50 μM, 24 h). I indicates time of addition of oligomycin, II of FCCP, and III of rotenone (mean±S.D.; n=3). (d) ATP measurements by luminescence after brefeldin A challenge (2.5 μM, 24 h) and CyPPA treatment (50 μM) (mean±S.D;. n=8; ***P<0.001 compared with untreated control). (e) TMRE fluorescence for detection of mitochondrial membrane potential analyzed by flow cytometry after 24 h brefeldin A (5 μM) with and without CyPPA pretreatment (50 μM, 24 h) (mean±S.D.; n=3; ***P<0.001 compared with untreated control). OCR, oxygen consumption rate
Intracellularly located SK2 channels are involved in CyPPA-mediated protection
SK channel activation was shown to inhibit N-methyl-d-aspartate-induced intracellular Ca2+ deregulation in differentiated neurons.30, 31, 32 Initial measurements of the [Ca2+]cyto levels (Figure 2a) could not distinguish between Ca2+ influx from extracellular space and the release from internal Ca2+ stores, for example, the ER or mitochondria. Therefore, we sought to differentiate between effects mediated by activation of SK2 channels located at the plasma membrane which may attenuate toxic Ca2+ influx from the extracellular space versus possible protective effects arising from the activation of SK2 channels located in intracellular compartments. Thus, we performed experiments in extracellular Ca2+-depleted medium and subsequently investigated HT-22 cell death. CyPPA reduced the ER stress-mediated cell death also in the absence of extracellular Ca2+ (Figure 6a and b) and largely inhibited brefeldin A-induced changes in cell morphology (Figure 6c). Furthermore, chelation of extracellular Ca2+ by EDTA did not affect the CyPPA-mediated protection (Figure 6d). Altogether, these results indicated that inhibition of detrimental Ca2+ influx from the extracellular space was not the main mechanism in CyPPA-mediated protection against ER stress-induced cell death.

Activation of intracellularly located SK2 channels is essential for CyPPA-mediated protection against ER stress. (a–c) HT-22 cells were challenged with brefeldin A (5 μM, 24 h) with and without CyPPA pretreatment (50 μM, 24 h) in the absence of extracellular calcium. (b) Quantification of AnnexinV and/or PI-positive cells (mean±S.D., n=3; ###P<0.001 compared with brefeldin A-only treated cells). (c) Representative pictures of HT-22 cell morphology after 24 h treatment in the absence of extracellular calcium. Scale bar 20 μm. (d) HT-22 cells were challenged with brefeldin A (5 μM, 24 h) with and without CyPPA pretreatment (50 μM, 24 h) in the presence of the Ca2+ chelator EDTA (2 mM); quantification of PI-positive cells (mean±S.D., n=3; ###P<0.001 compared with brefeldin A-only treated cells). (e) HT-22 cells were pretreated with CyPPA (50 μM, 24 h) followed by treatment with the SK2 channel blockers apamin (10 μM) and UCL1684 (50 μM) in conjunction with brefeldin A (5 μM) for 24 h. Quantification of AnnexinV/PI-positive cells (mean±S.D., n=3; ###P<0.001 compared with CyPPA pretreated and brefeldin A-only treated cells)
To further support the protective potential of intracellularly located SK2 channels we used different SK channel blockers in the presence or absence of brefeldin A. Apamin is a commonly used, highly selective peptide presumed to block SK channels only at the level of the plasma membrane because it has an extracellular binding site.14, 33 On the other hand, the non-peptide SK channel blocker UCL1684 is also capable of blocking intracellularly located SK channels.14, 34 In the present study, only UCL1684 completely abolished the protective effects of CyPPA, while apamin failed to affect CyPPA-mediated protection in the presence and absence of extracellular calcium (Figure 6e and Supplementary Figure S7).
SK2 channels are located at the ER membrane and prevent brefeldin A-induced ER Ca2+ depletion
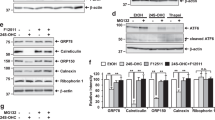
To further investigate intracellular localization of SK2 channels, we co-transfected HT-22 cells with ER-targeted red fluorescence protein to visualize ER and simultaneously with green fluorescent protein (GFP)-tagged SK2 channels. Merged panels visualize partial co-localization of ER marker and GFP-labeled SK2 channels, indicating SK2 channel residence in HT-22 ER membranes (Figure 7a). To further prove the topology of SK2 channels at the level of ER membranes, we performed subcellular fractionation analysis of HT-22 neurons. Besides plasma membrane and mitochondrial localization,18 the utilized subtype specific antibody identified SK2 channel also at the ER microsome-enriched protein fraction (Figure 7b and Supplementary Figure S8). These data demonstrate an intracellular residence of SK2 channels at the ER membrane of neuronal HT-22 cells, with potential importance for the CyPPA-mediated protective effects against ER stress.

SK2 channels are located in the ER membrane and reduce brefeldin A-induced ER calcium release. (a) HT-22 cells were transfected with ER-targeted red fluorescence protein plasmid to visualize ER (red fluorescence) and SK2 channel green fluorescence protein plasmid (green fluorescence). Co-localization between ER and SK2 channel in the representative histogram was determined for the area indicated by the dashed line in the overlay picture. Pearson’s correlation was calculated for various regions within one cell (n=6, mean±S.D.). (b) Subcellular fractionation of HT-22 cells shows SK2 channel localization in microsome-enriched fractions (ER) which are depleted from mitochondrial and cytoplasmic contamination, as shown by immunodetection of marker proteins. Equal amounts of protein were loaded for each fraction (12 μg; mitochondria (mito), cytosol (cyto), plasma membrane (PM), endoplasmic reticulum (ER)). (c–f) HT-22 cells were stimulated with brefeldin A (2.5 μM) for 24 h with or without CyPPA pretreatment (50 μM, 24 h). ER store depletion with thapisgargin (2 μM) was performed after 30 s basal measurement in EGTA buffer. (c) Representative pictures of [Ca2+]i intensity measured using Fura2 (340/380 nm ratio) before and 2 min after ER store depletion. (d) Representative kinetic profiles of [Ca2+]ER values (n≥10). (e) For quantification of [Ca2+]ER peak data were pooled from at least four wells for each condition, containing >40 cells. Peak values were normalized to basal [Ca2+]i intensity and presented as % of control (n=4, mean±S.D.; *P<0.05 compared with untreated control, #P<0.05 compared with brefeldin A-only treated cells, ANOVA, Scheffé’s test). (d) Data were pooled from at least five wells for each condition, containing >40 cells. The area under the curve (AUC) was calculated and presented as mean±S.D. in bar graphs (n=5; *P<0.05 compared with untreated control, #P<0.05 compared with brefeldin A-only treated cells). (g) HT-22 cells were pretreated with CyPPA (50 μM, 24 h) followed by treatment with quinine (25 μM) in conjunction with brefeldin A (5 μM) for 24 h. Quantification of AnnexinV/PI-positive cells (mean±S.D., n=3; ###P<0.001, n.s.P>0.05 compared with brefeldin A-only treated cells)
Decreased ER Ca2+ levels are associated with accumulation of unfolded proteins in the ER lumen and ER stress.35 Therefore, we studied the content of the [Ca2+]ER store by staining the cells with Fura2-AM that binds to free intracellular Ca2+, followed by treatment with the irreversible SERCA inhibitor thapsigargin for [Ca2+]ER store depletion. SK2 channel activation with CyPPA had no significant impact on basal [Ca2+]ER levels compared with untreated control cells, quantified after thapsigargin exposure as maximal Fura2-fluorescence (Figure 7d) as well as area under the curve for Fura2 fluorescence (AUC; Figure 7f). Furthermore, we performed long-term treatments with brefeldin A (24 h) and detected reduced [Ca2+]ER levels (Figure 7c–f). Interestingly, SK2 channel activation by CyPPA pretreatment prevented the brefeldin A-induced loss of [Ca2+]ER levels (Figure 7c–f).
Recent studies indicated that activity of the ER-located K+/H+ exchanger is required for [Ca2+]ER uptake. SK channels were presumed as a possible re-entry mechanism for the leaking potassium (K+), therefore also contributing to [Ca2+]ER uptake.13 To test this hypothesis in our model of ER stress, we applied quinine and propranolol, two known blockers of the ER membrane located K+/H+ exchanger,14 and identified significant inhibition of the protective effects of SK2 channel activation in ER stress-induced cell death (Figure 7g,Supplementary Figure S9).
Discussion
Our study demonstrates that activation of SK2 channels by CyPPA protects neuronal HT-22 cells against ER stress-induced apoptosis. We suggest that activation of SK2 channels located at the ER membrane (SK2ER) may prevent [Ca2+]ER depletion and resulting cellular demise under conditions of prolonged ER stress.
ER stress-caused alterations in intracellular calcium homeostasis contribute to cell death via different mechanisms. Depletion of the [Ca2+]ER stores disturbs protein folding inside the ER lumen, resulting in prolonged UPR activation and switching towards proapoptotic signaling.11 In the present study, the use of brefeldin A resulted in significantly reduced [Ca2+]ER stores in neuronal cells (Figure 7) and enhanced the expression of the UPR proteins XBP-1, ATF4, CHOP and GADD34 (Figure 3). These findings are in line with previous studies showing that long-term treatment with brefeldin A caused a moderate reduction in steady-state [Ca2+]ER levels in HeLA cells as well as in neuron-like NT-2 cells.36, 37 Furthermore, it was previously shown that elevated [Ca2+]cyto levels activated pro-caspase-12, thereby inducing the caspase signaling cascade independent of mitochondrial damage.19, 38 Our study demonstrates that cleavage of caspase-12 was induced by brefeldin A (Figure 2b and c), suggesting that the resulting apoptosis occurred independently of mitochondrial pathways of cell death. Similar to previous studies,37 we found that brefeldin A treatment affects mitochondria only following 24 h treatment. Therefore, mitochondrial demise is likely more a late consequence than a major key decision point in ER stress-induced cell death.
We provide strong evidence that SK2 channels are located at the ER of HT-22 cells (Figure 7). Furthermore, our results demonstrate major protective effects against ER stress by activation of intracellularly located SK2 channels (Figure 6). Therefore, we conclude that SK2ER channels are a major target of CyPPA-mediated protection against ER stress. Generally, K+ fluxes across the ER membrane are supposed to be important for [Ca2+]ER influx and release to balance charge movements.14, 39, 40 During the phase of [Ca2+]ER uptake, SERCA activity is associated with an extrusion of protons.41 Therefore, compensating mechanisms, responsible for proton re-entry, are thought to be necessary to prevent ER alkalization and enable Ca2+ uptake by the SERCA pump, which has been shown to be inoperable with increasing pH.42 Recently the K+/H+ exchanger was proposed as a possible executor of ER proton re-uptake and K+ release during Ca2+ uptake.14 Here, the SK channel activator CyPPA significantly attenuated brefeldin A-mediated [Ca2+]ER depletion. Blockers of the K+/H+ exchanger inhibited the protective effects of SK2 channel activation (Figure 7g and Supplementary Figure S9). In addition, cell death induced by the irreversible SERCA blocker thapsigargin could not be prevented by CyPPA (Supplementary Figure S2B). These results suggested that activation of SK2ER channels determined K+ entry from the cytoplasm into the ER lumen to allow for proton influx via the K+/H+ exchanger, thereby regulating Ca2+ uptake via SERCA and [Ca2+]ER retention (Figure 8).

Supposed mechanism of CyPPA-mediated protective effects. (a) In the absence of CyPPA brefeldin A mediates intracellular Ca2+ disturbances, activation of the UPR, mitochondrial demise and activation of the caspase cascade. (b) SK2ER channel activation diminishes the brefeldin A effects, facilitates intracellular Ca2+ homeostasis and increases cellular survival
In line with our previous studies,18, 29 SK channel activation elicits significant changes in mitochondrial function, including reduced basal respiration. Earlier studies indicate that neuronal apoptosis can be diminished by lowered mitochondrial respiration and ATP production.43, 44, 45, 46 Here, the inhibitory effects on mitochondrial respiration were accompanied by an initial boost in glycolysis (Supplementary Figure S6), which was previously associated with the protective properties of CyPPA pretreatment in oxidative damage.29 However, further investigations are needed to clarify whether changes in mitochondrial respiration by SK channel activation contribute to the protective effects under conditions of ER stress.
Maintenance of Ca2+ER homeostasis may play an important role in the cellular antioxidant defense mechanism.10 The increase in the [Ca2+]Cyto level after [Ca2+]ER store depletion, besides affecting the activation of caspase-12, might induce as a secondary effect a mitochondrial Ca2+ overload and ROS production. Moreover, decreases in [Ca2+]ER stimulate oxidative protein folding in ER to cope with the increasing amount of unfolded proteins. These energy-dependent processes lead to an ATP depletion, mitochondrial oxidative phosphorylation and thus ROS production.10 Here, the SK channel activator was able to partly reduce ROS formation and to prevent the moderate increase in [O2−]m production only under prolonged (24 h) ER stress (Figure 4). However, we conclude that the detected effects of CyPPA on mitochondrial morphology, function and bioenergetics are neither necessary nor sufficient to prevent ER stress-induced cell death.
Here, splicing of XBP-1 mRNA and enhanced expression of ATF4 and GADD34 was prevented by the use of CyPPA (Figure 3). Previous findings demonstrated a correlation of the intracellular Ca2+ homeostasis and the activation of the UPR.2 For example, in neuronal PC12 cells the activation of the UPR was enhanced when Ca2+ER store depletion was intensified by the use of BAPTA-AM in a model of thapsigargin-induced ER stress.11 Further, the carbazole derivative 16-14 [9-(3-cyanobenzyl)-1,4-dimethylcarbazole], which was able to maintain PC12 cellular Ca2+ homeostasis by compensation of the thapsigargin-induced [Ca2+]ER release, also attenuated the UPR after ER stress stimuli.12 Therefore, the modulating effects of CyPPA pretreatment on the different transcription factors linked to the UPR may also be attributed to the stabilization of intracellular Ca2+ homeostasis.
Surprisingly, the combinatory treatment of brefeldin A and CyPPA had synergistic effects on CHOP protein levels (Figure 3c). Recent studies showed that, besides its role in promoting cell death,22 CHOP could also inhibit apoptosis, suggesting a complex role of this transcription factor in certain cases.47, 48, 49 In our model of ER stress, CHOP knockdown had only minor effects on apoptosis and it did not affect the protective potential of CyPPA (Figure 3e). Therefore, we conclude that in our experimental settings enhanced expression of CHOP alone is not sufficient to promote cell death, but it is also not required for CyPPA-mediated protection. Recent studies demonstrated that adenovirus-mediated delivery of CHOP did not affect mouse embryonic fibroblast cell viability but enhanced expression of ATF4 decreased cell survival.50 ATF4 directly binds to and trans-activates a conserved ATF site in the GADD34 promoter during ER stress,51 resulting in the resumption of general translation and apoptosis.52, 53 This is in line with our present finding that knockdown of ATF4, but not CHOP, protected against brefeldin A-induced cell death (Figure 3). It still remains unclear, how SK2 channel activation can exhibit diverse effects on sXBP1 and ATF4 on one hand, and CHOP proteins on the other. Previous studies revealed that CHOP can also be induced by different mechanisms, such as, for example, proteolysis of ATF6 or the induction of ATF2.54, 55 Therefore, SK channel activation may initiate additional mechanisms that cause increased CHOP expression, independent of ATF4 and sXBP1. In fact, the additive effects of brefeldin A and CyPPA on CHOP protein levels support the conclusion that different pathways of protein regulation were involved.
Taken together, our study suggests a substantial role of SK2 channels in the regulation of [Ca2+]ER uptake and retention with neuroprotective properties under ER stress conditions.
Materials and Methods
Cell culture
HT-22 cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine (all from PAA, Cölbe, Germany). One day after seeding, cells were pretreated with CyPPA 50 μM (Sigma-Aldrich, Munich, Germany) for 24 h. Afterwards the cells were treated with different concentrations of brefeldin A, 0.25 μM thapsigargin or 0.025 μg/ml tunicamycin (all from Sigma-Aldrich). Additional substances used were 25–100 μM NS309 (Sigma-Aldrich), 125 μM 1-EBIO (Sigma-Aldrich), 50 μM Q-VD-OPh (Merck Millipore, Darmstadt, Germany), 10 μM apamin (Latoxan, Valence, France), 50 μM UCL1684 (Tocris Bioscience, Bristol, UK), 25 μM quinine (Sigma-Aldrich) or 25 μM propranolol (Sigma-Aldrich) in DMEM with or without calcium.
Cell viability assay
Cell viability was investigated in a 96-well plate format using the colorimetric MTT reduction assay. MTT solution (0.5 mg/ml) (Sigma-Aldrich) was applied to the cells for 1 h. Afterwards, the MTT containing medium was entirely removed and incubated for 1 h at −80 °C. After adding DMSO for dye solution and incubation for 1 h at 37 °C, the absorbance was determined with a FLUOstar Optima reader (BMG Labtech, Offenburg, Germany) at 570 nm with a reference filter at 630 nm. Untreated control was regarded as 100% cell viability.
Cell death assays
For AnnexinV/PI double staining, the cells were cultured in a 24-well plate format and harvested by using Trypsin/EDTA after treatment. Cells were incubated in binding buffer containing AnnexinV-FITC and propidium iodide (PI) (PromoKine, Heidelberg, Germany) at room temperature for 10 min. Flow cytometric analysis was performed with excitation at 488 nm and emission at 525/530 nm for AnnexinV and at 690/50 nm for PI using the Guava Easy Cyte 6–2 L system (Merck Millipore). Dead cells were quantified from AnnexinV and AnnexinV/ PI-positive cells. Data were collected from 10 000 cells per condition.
Microscopic analysis
Microscopy pictures of cell morphology were performed using a Leica DM 6000 microscope and LAS AF software (Leica Microsystems, Wetzlar, Germany).
Measurements of cytosolic calcium
After treatment in black 96-well plates, cells were incubated with 1 μM Fluo-4 AM, 0.25 μM probenecid and 0.2% pluronic F-127 (all from Invitrogen) in HEPES-Ringer buffer (136.4 mM NaCl, 5.6 mM KCl, 1 mM MgCl2, 2.2 mM CaCl2, 10 mM HEPES, 5 mM glucose, 0.1% BSA, pH 7.4) for 1 h. Afterwards, Fluo-4 AM containing solution was removed and cells were incubated for an additional 30 min in HEPES-Ringer buffer with 0.25 μM probenecid. Fluorescence was excited at 485 nm; emission was detected at 520 nm (FLUOstar Optima Reader; BMG Labtech).
Western blotting analysis of ER stress markers
Cell extracts were prepared from cells lysed in a buffer containing 50 mM tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton-X-100, 0.5% sodium-deoxycholate, 1 mM PMSF, 1.25 mM Na4P2O7 and 2 mM Na3VO4. The cellular suspension was frozen/thawed three times in liquid nitrogen and centrifuged with 10 000 r.p.m., 10 min at 4 °C. Protein concentration in the supernatant was measured using Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). Samples were denatured at 95 °C for 5 min in sample buffer containing 40% glycerine, 312.5 mM tris/HCl pH 6.8, 8% SDS, 20% β-mercaptoethanol, 0.08% bromophenol blue. Equal amounts of each protein sample were separated by electrophoresis on 12.5% SDS-polyacrylamide gel. Proteins were electroblotted on PVDF membranes and incubated overnight at 4 °C with one of the following primary antibodies: mouse monoclonal anti-CHOP (1 : 500), goat polyclonal anti-ATF4 (1 : 500), mouse polyclonal anti-GADD34 (1 : 1000) (all from Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit monoclonal anti-caspase-3 (1 : 1000), rabbit polyclonal anti-caspase-12 (1 : 1000), rabbit polyclonal anti-α/β-tubulin (1 : 10 000) (all from Cell Signalling Technology Inc., Danvers, MA, USA). After washing, membranes were incubated with peroxidase-conjugated secondary antibodies (1 : 2500) (Vector Labratories, Peterborough, UK) for 1 h at room temperature. Bands were detected on a ChemiDoc XRS system (Bio-Rad Laboratories, Munich, Germany) after incubation with HRP-Juice (PJK, Kleinbittersdorf, Germany) for 2–5 min. Quantification was performed using Image Lab software 4.0 (Bio-Rad Laboratories).
Transfection of siRNA
Reverse transfection was performed in 24-well plates by incubating the siRNAs (40 nM) with Lipofectamine RNAiMAX in Opti-MEM (Invitrogen) for 15 min at room temperature. In all, 40 000 cells/well were added in media without antibiotics and treated 24 h after transfection. Following siRNAs (Eurofins MWG Operon, Ebersberg, Germany) were used:
CHOP: 5′-UGUUUCCGUUUCCUAGUUCUU-3′18
ATF4: 5′-AUCGAAGUCAAACUCUUUCUU-3′18
Non-specific (Scr): 5′-UAAUGAAUUGGAACGCAUATT-3′18
RT-PCR analysis
Total RNA was extracted using inviTrap Spin Universal RNA Mini Kit (Stratec Molecular, Berlin, Germany) following the manufacturer’s protocol. Reverse transcriptase reactions were performed with SuperScript III RT/Platinum Taq Mix (Invitrogen) in a thermo cycler with settings at 42 °C for 30 min. Following primers (Eurofins MWG Operon) were used:
Murine XBP-1: Forward: 5′-GAACCAGGAGTTAAGAACACG-3′;18
Reverse: 5′-AGGCAACAGTGTCAGAGTCC-3′17
Murine ATF4: Forward: 5′-AUCGAAGUCAAACUCUUUCUU-3′;18
Reverse: 5′-AAGAAAGAGUUUGACUUCGAU-3′18
Murine CHOP: Forward: 5′-CATACACCACCACACCTGAAAG-3′;19
Reverse: 5′-CCGTTTCCTAGTTCTTCCTTGC-3′19
The amplification of cDNA was carried out at the following steps: (a) denaturing at 95 °C for 4 min; (b) 94 °C for 30 s (XBP-1 and ATF4), or 15 s (CHOP); (c) annealing at 58 °C for 30 s for XBP-1, annealing at 59 °C for 1 min for ATF4, annealing at 55 °C for 30 s for CHOP; (d) extension at 72 °C for 30 s for XBP-1 and ATF4; at 68 °C for 45 s for CHOP; 30 cycles for XBP-1 and ATF4, and 26 cycles for CHOP were performed with steps (b)–(d). The final extension step was set to 72 °C (XBP-1 and ATF4) or 58 °C (CHOP) for 5 min.
Detection of oxidative stress
Assays for intracellular oxidative stress were performed by flow cytometry using the Guava Easy Cyte 6–2 L system (Merck Millipore). The fluorescence was always excited using a 488 nm UV line argon laser. Data were collected from at least 5000 cells.
For measurements of the cytosolic ROS concentration CM-H2DCFDA (Invitrogen), the reduced and acetylated form of DCF was used. Cells were loaded with 2.5 μM dye in serum-deprived medium for 30 min, followed by 30 min incubation without dye in FCS-containing DMEM. The fluorescence was detected at 525/530 nm using the green filter.
For detection of the mitochondrial superoxide concentration [O2−]m cells were incubated for 30 min with 2.5 μM MitoSOX red (Invitrogen). The fluorescence was detected at 690/50 nm using the red filter.
Mitochondrial morphology
Cells were incubated with 250 μM MitoTracker Deep Red (Sigma-Aldrich) for 30–40 min. After fixation with paraformaldehyde, the red fluorescence was excited at 620±60 nm and emission was detected at 700±75 nm. For each condition 500 cells were counted. Mitochondrial morphology was assessed and classified into three different categories. Category I are elongated, tubulin-like mitochondria, category II large-fragmented mitochondria, and category III represents cells with small-fragmented mitochondria which are located only around the nucleus. Representative images were taken by a confocal laser scanning microscope (Leica SP5; Leica, Wetzlar, Germany).
Measurement of cellular oxygen consumption rate and extracellular acidification rate
Oxygen consumption rate measurements were carried out using different agents (Oligomycin, FCCP, Rotenone and Antimycin A) from the Mitochondrial Stress Kit, as previously described with minor modifications.44 Briefly, HT-22 cells were seeded in XF96-well cell culture microplates (Seahorse Bioscience, North Billerica, MA, USA) at 10 000 cells/well in 4.5 g/l glucose-containing media and incubated at 37 °C and 5% CO2 for ~20 h. Before the measurements were made, the growth medium was washed off and replaced with ~180 μl of assay medium (with 4.5 g/l glucose as the sugar source, 2 mM glutamine, 1 mM pyruvate, pH 7.35) and cells were incubated at 37 °C for 60 min. Three baseline measurements were recorded before the addition of compounds. Oligomycin (20 μl) was injected at a final concentration of 3 μM, FCCP (22.5 μl) at a concentration of 0.4 μM and Rotenone/Antimycin A (25 μl) at a concentration of 1 μM. Three measurements were performed after the addition of each compound (4 min mixing followed by 3 min measuring).
Regarding extracellular acidification rate (ECAR) measurements, three baseline measurements were recorded before the addition of vehicle or CyPPA (50 μM). The measurements were performed in assay medium supplemented with 4.5 g/l glucose as the sugar source, 2 mM glutamine, 1 mM pyruvate, pH 7.35. ECAR values were recorded for 6 h (4 min mixing followed by 3 min measuring, with a waiting interval of 15 min between measurements).
ATP measurements
Determination of ATP levels was performed by ViaLightTM plus kit (Lonza, Walskerville, MI, USA) in luminescence compatible 96-well plates according to the manufacturer’s protocol. Luminescence was measured with a FLUOstar Optima reader (BMG Labtech).
Measurement of ΔΨm
Loss of the ΔΨm was determined by TMRE (tetramethylrhodamine, ethyl ester, perchlorate) fluorescent dye (Sigma-Aldrich). The cells were harvested with trypsin/EDTA and incubated for 30 min with 0.2 μM TMRE. CCCP (50 μM) was used as a positive control. The assay was performed with flow cytometric analysis using the Guava Easy Cyte 6–2 l system (Merck Millipore). The fluorescence was excited at 488 nm and emission was detected at 690/50 nm using the red filter. Data were collected from 5000 cells each condition.
Plasmid transfection and immunostaining
For analysis of SK2 channel localization, HT-22 cells were seeded on cover slips (25 mm diameter) in a six-well plate format at a density of 120 000 cells. HT-22 cells were transfected with ER-targeted red fluorescence protein (kindly provided by Prof. Erik Snapp) and GFP-tagged SK2 channel (kindly provided by Prof. Bernd Fakler) using an attractene transfection protocol (Qiagen, Hilden, Germany). Forty-eight hours after transfection cells were fixated with 4% paraformaldehyde. Images were acquired using a confocal laser scanning microscope (Leica SP5; Leica). SK2-GFP fluorescence was excited at 488 nm, and emissions were detected between 500 and 535 nm bandwidth. Red fluorescence was detected by excitation at 633 nm and emission between 640 and 750 nm. Co-localization was assessed with ImageJ.
Subcellular fractionation analysis
Subcellular fractionation was performed as previously described56 with minor modifications. Briefly, confluent HT-22 cells from 25 flasks (75 cm2) were collected by trypsinization. After washing, the cell pellet was resuspended in a buffer containing 225 mM mannitol, 75 mM sucrose and 30 mM Tris-HCl (pH 7.4) and homogenized with a Potter-Elvehjem homogenizer. The homogenate was centrifuged at 800 × g for 5 min to remove unbroken cells and nuclei. The supernatant was further centrifuged at 10 000 × g for 10 min to pellet the mitochondria. The resulting supernatant was centrifuged at 25 000 × g for 20 min to pellet the plasma membrane. For separation of microsomes and cytosolic proteins the resulting supernatant was centrifuged at 95 000 × g for 2.5 h. Equal amounts of each protein sample were separated by electrophoresis on a 4–12.5% Bis-Tris gradient gel (Life Technologies, Carlsbad, CA, USA). Proteins were electroblotted on nitrocellulose membranes and incubated overnight at 4 °C with one of the following primary antibodies: rabbit polyclonal anti-SK2 (1 : 1000, kindly provided by Prof. Hans-Günther Knaus), rabbit monoclonal anti-PERK (1 : 1000; Cell Signalling Technology Inc.), mouse anti-Tim23 (BD Pharmingen, Franklin Lakes, NJ, USA), goat polyclonal anti-Actin (Santa Cruz Biotechnology Inc.). After washing, membranes were incubated with peroxidase-conjugated secondary antibodies (1 : 2500–1 : 5000) for 1 h at room temperature. For detection, membranes were incubated with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposed to an autoradiographic film (CL-Xposure Film; Thermo Scientific).
Life cell imaging of ER calcium release
HT-22 cells were seeded on cover slips (25 mm diameter) in a six-well plate format at a density of 60 000 cells. After treatment, cells were incubated with 2 μM Fura2-AM for 30 min at room temperature in HEPES-Ringer buffer (HRB; 136.4 mM NaCl, 5.6 mM KCl, 1 mM MgCl2, 2.2 mM CaCl2, 10 mM HEPES, 5 mM glucose, 0.1% BSA, pH 7.4). Afterwards, the dye was removed and cells were incubated for an additional 15 min at room temperature in HRB. EGTA and thapsigargin were diluted in HRB without calcium. Basal calcium was measured in HRB without calcium for 30 s before application of thapsigargin/EGTA. Images were acquired using a Polychrome II monochromator and an IMAGO CCD camera (Till Photonics, Martinsried, Germany) coupled with an inverted microscope (IX70; Olympus, Hamburg, Germany). Images were collected using a 20 × 0.8 numerical aperture oil immersion objective. An increase in intracellular Ca2+ was reflected by a fluorescence increase when exciting at 340 nm and a corresponding decrease with excitation at 380 nm. F340 and F380 were recorded separately and combined (fluorescence ratio: r=F340/F380) after background subtraction (fluorescence of a cell-free area).
Statistical analysis
Multiple comparisons were performed by analysis of variance (ANOVA) followed by Scheffé's post hoc test. P<0.05 was used as a threshold P-value for definition of significance. Calculations were performed with the Winstat standard statistical software (R. Fitch Software, Bad Krozingen, Germany) package.
Abbreviations
- [Ca2+]cyto:
-
cytoplasmic calcium
- [Ca2+]ER:
-
ER calcium
- [O2−]m:
-
mitochondrial superoxides
- 1-EBIO:
-
1-ethylbenzimidazolinone
- ATF4:
-
activating transcription factor 4
- ATF6:
-
activating transcription factor 6
- ATP:
-
adenosine triphosphate
- AUC:
-
area under the curve
- Ca2+:
-
calcium ion
- CHOP:
-
CCAAT/enhancer-binding protein homologous protein
- CM-H2DCFDA:
-
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- CyPPA:
-
cyclohexyl-[2E-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine
- ECAR:
-
extracellular acidification rate
- ER stress:
-
stress of the endoplasmic reticulum
- ER:
-
endoplasmic reticulum
- FCCP:
-
carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- FITC:
-
fluorescein isothiocyanate
- Fura2-AM:
-
Fura2-acetoxymethyl ester
- GADD34:
-
growth arrest and DNA damage-inducible protein 34
- GFP:
-
green fluorescent protein
- grp78:
-
78 kDa glucose-regulated protein
- H+:
-
proton
- HRB:
-
HEPES-Ringer buffer
- HT-22 cells:
-
immortalized mouse hippocampal neurons
- IRE1:
-
inositol-requiring enzyme
- K+:
-
potassium ion
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NS309:
-
6,7-dichloro-1H-indole-2,3-dione 3-oxime
- PERK:
-
PRKR-like endoplasmic reticulum kinase
- PI:
-
propidium iodide
- Q-VD-OPh:
-
N-(2-quinolyl)-l-valyl-l-aspartyl-(2,6-difluorophenoxy) methylketone
- ROS:
-
reactive oxygen species
- RT-PCR:
-
reverse transcription polymerase chain reaction
- SERCA:
-
sarco/endoplasmic reticulum Ca2+-ATPase
- siRNA:
-
small interfering RNA
- SK channel:
-
small-conductance calcium-activated potassium channel
- SK2 channel:
-
small-conductance calcium-activated potassium channel, subtype 2
- SK2ER:
-
ER-located SK2 channels
- SK3 channel:
-
small-conductance calcium-activated potassium channel, subtype 3
- TMRE:
-
tetramethylrhodamine, ethyl ester, perchlorate
- UCL1684:
-
6,12,19,20,25,26-hexahydro-5,27:13,18:21,24-trietheno-11,7-metheno-7H-dibenzo [b,n] [1,5,12,16]tetraazacyclotricosine-5,13-diium dibromide
- UPR:
-
unfolded protein response
- XBP-1:
-
X-box binding protein 1
- ΔΨm:
-
mitochondrial membrane potential
References
Hebert DN, Molinari M . In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev 2007; 87: 1377–1408.
Ma Y, Hendershot LM . ER chaperone functions during normal and stress conditions. J Chem Neuroanat 2004; 28: 51–65.
Mekahli D, Bultynck G, Parys JB, de Smedt H, Missiaen L . Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 2011; 3: a004317.
Schroder M, Kaufman RJ . The mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–789.
Sano R, Reed JC . ER stress-induced cell death mechanisms. Biochim Biophys Acta 2013; 1833: 3460–3470.
Matus S, Glimcher LH, Hetz C . Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol 2011; 23: 239–252.
Kraskiewicz H, Fitzgerald U . Interfering with endoplasmic reticulum stress. Trends Pharmacol Sci 2012; 33: 53–63.
Qi X, Okuma Y, Hosoi T, Nomura Y . Edaravone protects against hypoxia/ischemia-induced endoplasmic reticulum dysfunction. J Pharmacol Exp Ther 2004; 311: 388–393.
Takano K, Kitao Y, Tabata Y, Miura H, Sato K, Takuma K et al. A dibenzoylmethane derivative protects dopaminergic neurons against both oxidative stress and endoplasmic reticulum stress. Am J Physiol Cell Physiol 2007; 293: C1884–C1894.
Malhotra JD, Kaufman RJ . Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 2007; 9: 2277–2293.
Yoshida I, Monji A, Tashiro K, Nakamura K, Inoue R, Kanba S . Depletion of intracellular Ca2+ store itself may be a major factor in thapsigargin-induced ER stress and apoptosis in PC12 cells. Neurochem Int 2006; 48: 696–702.
Miura H, Takano K, Kitao Y, Hibino S, Choshi T, Murakami R et al. A carbazole derivative protects cells against endoplasmic reticulum (ER) stress and glutathione depletion. J Pharmacol Sci 2008; 108: 164–171.
Kuum M, Veksler V, Kaasik A . Potassium fluxes across the endoplasmic reticulum and their role in endoplasmic reticulum calcium homeostasis. Cell Calcium 2014; 58: 79–85.
Kuum M, Veksler V, Liiv J, Ventura-Clapier R, Kaasik A . Endoplasmic reticulum potassium-hydrogen exchanger and small conductance calcium-activated potassium channel activities are essential for ER calcium uptake in neurons and cardiomyocytes. J Cell Sci 2012; 125 (Pt 3): 625–633.
Maher P, Davis JB . The role of monoamine metabolism in oxidative glutamate toxicity. J Neurosci 1996; 16: 6394–6401.
Klausner RD, Donaldson JG, Lippincott-Schwartz J, Brefeldin A . Insights into the control of membrane traffic and organelle structure. J Cell Biol 1992; 116: 1071–1080.
Hougaard C, Eriksen BL, Jorgensen S, Johansen TH, Dyhring T, Madsen LS et al. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br J Pharmacol 2007; 151: 655–665.
Dolga AM, Netter MF, Perocchi F, Doti N, Meissner L, Tobaben S et al. Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J Biol Chem 2013; 288: 10792–10804.
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403: 98–103.
Demaurex N, Frieden M, Arnaudeau S ER Calcium and ER Chaperones: New Players in Apoptosis? Madame Curie Bioscience Database, Landes Bioscience, 2000.
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y . An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 2002; 277: 34287–34294.
Rao RV, Ellerby HM, Bredesen DE . Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ 2004; 11: 372–380.
Hetz C, Chevet E, Harding HP . Targeting the unfolded protein response in disease. Nat Rev Drug Discov 2013; 12: 703–719.
Bhandary B, Marahatta A, Kim H, Chae H . An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 2012; 14: 434–456.
Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 2011; 124 (Pt 13): 2143–2152.
Vannuvel K, Renard P, Raes M, Arnould T . Functional and morphological impact of ER stress on mitochondria. J Cell Physiol 2013; 228: 1802–1818.
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E . Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 2008; 9: 505–518.
Grohm J, Kim S, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J et al. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ 2012; 19: 1446–1458.
Richter M, Nickel C, Apel L, Kaas A, Dodel R, Culmsee C et al. SK channel activation modulates mitochondrial respiration and attenuates neuronal HT-22 cell damage induced by HO. Neurochem Int 2015; 81: 63–75.
Dolga AM, Terpolilli N, Kepura F, Nijholt IM, Knaus H, D'Orsi B et al. KCa2 channels activation prevents Ca2+i deregulation and reduces neuronal death following glutamate toxicity and cerebral ischemia. Cell Death Dis 2011; 2: e147.
Allen D, Nakayama S, Kuroiwa M, Nakano T, Palmateer J, Kosaka Y et al. SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J Cereb Blood Flow Metab 2011; 31: 2302–2312.
Bond CT, Maylie J, Adelman JP . SK channels in excitability, pacemaking and synaptic integration. Curr Opin Neurobiol 2005; 15: 305–311.
Faber ES . Functions and modulation of neuronal SK channels. Cell Biochem Biophys 2009; 55: 127–139.
Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH . Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)-diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca(2+)-activated K+ channel. J Med Chem 1998; 41: 2–5.
Paschen W . Dependence of vital cell function on endoplasmic reticulum calcium levels: implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium 2001; 29: 1–11.
Chami M, Oules B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Brechot P . Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell 2008; 32: 641–651.
Arduino DM, Esteves AR, Domingues AF, Pereira CM, Cardoso SM, Oliveira CR . ER-mediated stress induces mitochondrial-dependent caspases activation in NT2 neuron-like cells. BMB Rep 2009; 42: 719–724.
Szegezdi E, Fitzgerald U, Samali A . Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci 2003; 1010: 186–194.
Ng K, Schwarzer S, Duchen MR, Tinker A . The intracellular localization and function of the ATP-sensitive K+ channel subunit Kir6.1. J Membr Biol 2010; 234: 137–147.
Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M et al. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 2007; 448: 78–82.
Yu X, Carroll S, Rigaud JL, Inesi G . H+ countertransport and electrogenicity of the sarcoplasmic reticulum Ca2+ pump in reconstituted proteoliposomes. Biophys J 1993; 64: 1232–1242.
Peinelt C, Apell H . Kinetics of the Ca(2+), H(+), and Mg(2+) interaction with the ion-binding sites of the SR Ca-ATPase. Biophys J 2002; 82 (1 Pt 1): 170–181.
Poltl D, Schildknecht S, Karreman C, Leist M . Uncoupling of ATP-depletion and cell death in human dopaminergic neurons. Neurotoxicology 2012; 33: 769–779.
Gohil VM, Offner N, Walker JA, Sheth SA, Fossale E, Gusella JF et al. Meclizine is neuroprotective in models of Huntington's disease. Hum Mol Genet 2011; 20: 294–300.
Eguchi Y, Shimizu S, Tsujimoto Y . Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 1997; 57: 1835–1840.
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P . Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 1997; 185: 1481–1486.
Engel T, Sanz-Rodgriguez A, Jimenez-Mateos EM, Concannon CG, Jimenez-Pacheco A, Moran C et al. CHOP regulates the p53-MDM2 axis and is required for neuronal survival after seizures. Brain 2013; 136 (Pt 2): 577–592.
Chen C, Wu C, Chiang C, Liao B, Liu S . C/EBP homologous protein (CHOP) deficiency aggravates hippocampal cell apoptosis and impairs memory performance. PLoS One 2012; 7: e40801.
Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ . The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem 2010; 285: 21329–21340.
Han J, Back SH, Hur J, Lin Y, Gildersleeve R, Shan J et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013; 15: 481–490.
Ma Y, Hendershot LM . Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem 2003; 278: 34864–34873.
Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–3077.
Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB et al. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis 2013; 4: e754.
Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 2000; 20: 6755–6767.
Bruhat A, Jousse C, Carraro V, Reimold AM, Ferrara M, Fafournoux P . Amino acids control mammalian gene transcription: activating transcription factor 2 is essential for the amino acid responsiveness of the CHOP promoter. Mol Cell Biol 2000; 20: 7192–7204.
Suski JM, Lebiedzinska M, Wojtala A, Duszynski J, Giorgi C, Pinton P et al. Isolation of plasma membrane-associated membranes from rat liver. Nat Protoc 2014; 9: 312–322.
Acknowledgements
We thank Katharina Elsässer for her excellent technical support and Emma Jane Esser for careful reading and corrections of the manuscript. This work was supported in part by a grant from the DFG (DO1525/3-1).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by S Kaufmann
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Richter, M., Vidovic, N., Honrath, B. et al. Activation of SK2 channels preserves ER Ca2+ homeostasis and protects against ER stress-induced cell death. Cell Death Differ 23, 814–827 (2016). https://doi.org/10.1038/cdd.2015.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.146
This article is cited by
-
Apamin as a BBB Shuttle and Its Effects on T Cell Population During the Experimental Autoimmune Encephalomyelitis-Induced Model of Multiple Sclerosis
Neurotoxicity Research (2021)
-
Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes
Acta Neuropathologica Communications (2019)
-
Translocation Associated Membrane Protein 1 Contributes to Chronic Constriction Injury-Induced Neuropathic Pain in the Dorsal Root Ganglion and Spinal Cord in Rats
Journal of Molecular Neuroscience (2018)
-
SK2 channels regulate mitochondrial respiration and mitochondrial Ca2+ uptake
Cell Death & Differentiation (2017)
