
Abstract
Despite being the most evolutionarily conserved of the mammalian caspases, little is understood about the cellular function of caspase-2 in normal tissues or what role caspase-2 may have in the progression of human disease. It has been reported that deletion of the caspase-2 gene (Casp2), accelerates Eμ-myc lymphomagenesis in mice, and thus caspase-2 may act as a tumor suppressor in hematological malignancies. Here, we sought to extend these findings to epithelial cancers by examining the potential role of caspase-2 as a tumor suppressor in the mouse mammary carcinogenesis model; MMTV/c-neu. The rate of tumor acquisition was significantly higher in multiparous Casp2−/−/MMTV mice compared with Casp2+/+/MMTV and Casp2+/−/MMTV mice. Cells from Casp2−/−/MMTV tumors were often multinucleated and displayed bizarre mitoses and karyomegaly, while cells from Casp2+/+/MMTV and Casp2+/−/MMTV tumors never displayed this phenotype. Tumors from Casp2−/−/MMTV animals had a significantly higher mitotic index than tumors from Casp2+/+/MMTV and Casp2+/−/MMTV animals. Cell cycle analysis of Casp2−/− E1A/Ras-transformed mouse embryonic fibroblasts (MEF) also indicated a higher proliferative rate in the absence of caspase-2. In vitro assays further illustrated that MEF had increased genomic instability in the absence of caspase-2. This appears to be due to disruption of the p53 pathway because we observed a concomitant decrease in the induction of the p53 target genes, Pidd, p21 and Mdm2. Thus caspase-2 may function as a tumor suppressor, in part, through regulation of cell division and genomic stability.
Similar content being viewed by others


Main
Caspase-2 is the most evolutionarily conserved of all the caspases, but its role in apoptosis remains unclear.1 Recent evidence suggests that caspase-2 may have a role in the suppression of tumorigenesis. Loss of caspase-2 in a mouse model of lymphoma, induced by transgenic c-Myc on the μ-enhancer (Eμ-Myc), greatly accelerated tumor growth.2 Furthermore, caspase-2-deficient mouse embryonic fibroblasts (MEF) showed evidence of enhanced transforming potential in vitro and formed more accelerated tumors in athymic nude mice.2
One of the hallmarks of tumorigenesis is that cancer cells avoid apoptosis, thus caspase-2 may suppress tumors by inducing apoptosis of potential tumor cells. Caspase-2 has been implicated in apoptosis induced by numerous stimuli. Caspase-2-deficient cells have been shown to be more resistant to apoptosis induced by heat shock,3 γ-irradiation,2 cytoskeletal disruptors such as vincristine,4 and neuronal stresses such as β-amyloid and growth factor withdrawal.5, 6 In addition, apoptosis induced by inhibition of the Chk1 pathway or the pentose phosphate pathways appears to require caspase-2.7, 8
Caspase-2 may also have a role in regulating cell cycle progression. Casp2−/− MEF proliferate at higher rates and caspase-2-deficient cells are resistant to DNA damage-induced cell cycle arrest.2, 9 In addition, caspase-2 is repressed during mitosis by phosphorylation on Serine 340 by cdk-cyclin B1.10 Therefore, caspase-2 may function as a tumor suppressor, in part, through regulation of cell division.
Caspase-2 does not appear to be frequently deleted in human tumors (oncomine.org, sanger.ac.uk/genetics/cgp). However, it has been demonstrated that caspase-2 expression is decreased in in vitro drug-resistant childhood leukemia subtypes such as T-ALL and AML.11 It has also been suggested that high levels of inactive procaspase-2 in the peripheral blood of patients with AML and ALL compared with controls may be correlated with decreased survival and poor prognosis.12, 13, 14 It is unclear if loss or reduced expression of caspase-2 accelerates the acquisition or progression of other types of tumors, or if the tumor suppressive function of caspase-2 is restricted to hematological cancers. Therefore, we sought to extend these previous observations to an epithelial cancer to determine if the tumor suppressive function of caspase-2 is more general.
Results
Lack of caspase-2 accelerates mammary tumor formation in MMTV/c-neu multiparous mice
To explore the role of caspase-2 in epithelial cancers, we used the MMTV/c-neu-murine model of breast cancer. MMTV/c-neu expresses an activated form of the rat Erbb2 (c-neu) oncogene due to a V664E substitution under the transcriptional control of the mouse mammary tumor virus (MMTV) promoter.15 This induces active c-neu expression in the entire mammary epithelium and drives mammary tumorigenesis.
We generated female Casp2+/+/MMTV, Casp2+/−/MMTV and Casp2−/−/MMTV mice in crosses that generated all three genotypes. This experiment was carried out in a mixed genetic background; therefore the controls for each Casp2−/−/MMTV and Casp2+/−/MMTV strain were the Casp2+/+/MMTV littermates of the experimental mice. In the MMTV/c-neu mouse model, multiparous females acquire tumors earlier than virgin females, likely due to the contribution of steroid hormones on the MMTV promoter in addition to increased rounds of cell proliferation and cell death involved with growth and involution of the mammary tissue that occur with each pregnancy.16 Therefore, we monitored the development of tumors in mice that were virgins (nulliparous) or had more than two litters (multiparous).
Genetic deletion of Casp2 significantly accelerated the rate of tumor formation in multiparous female animals compared with Casp2+/+/MMTV multiparous female animals (Figure 1a). The loss of one copy of Casp2 was sufficient to partially accelerate tumor acquisition compared with Casp2+/+/MMTV animals but this was not statistically significant (P=0.0521, compared with wild-type). In mice lacking both copies of Casp2, the acceleration in tumor acquisition was most pronounced and statistically significant (P=0.0020, compared with wild-type). The median tumor-free survival of Casp2−/−/MMTV multiparous animals was 196 days, compared with 223 days for Casp2+/−/MMTV animals and 241 days for Casp2+/+/MMTV animals (Figure 1a). The incidence of tumor acquisition was ∼70% after 400 days for Casp2+/+/MMTV animals, consistent with published results.15 In contrast, 100% of the multiparous Casp2+/−/MMTV and multiparous Casp2−/−/MMTV females acquired tumors by day 271 and 255, respectively (Figure 1a). Parallel examination of the nulliparous MMTV animals revealed no differences in the rate or incidence of tumor acquisition in Casp2+/+/MMTV, Casp2+/−/MMTV or Casp2−/−/MMTV animals (Figure 1b), suggesting that the tumor suppressive role of caspase-2 may be restricted to tissues that have high proliferative rates.

Genetic deletion of Casp2 accelerates MMTV/c-neu-driven tumorigenesis in multiparous mice. Casp2+/+/MMTV, Casp2+/−/MMTV and Casp2−/−/MMTV multiparous (a) or nulliparous (b) female mice were monitored for the appearance of mammary tumors every second day over the indicated time period. The percentage of animals that were not moribund is shown. Mice were considered ‘not moribund’ until they either required euthanization due to illness or immobilization as a result of their tumor burden. When animals became moribund, mice were submitted for necropsy and routine histology. The P-values shown compare all three genotypes and were determined by Log-Rank (Mantel-Cox) test. The median survival for multiparous mice was 241 days (Casp2+/+/MMTV) and 196 days (Casp2−/−/MMTV). The median survival for nulliparous mice was 321 days (Casp2+/+/MMTV) and 306.5 days (Casp2−/−/MMTV)
In MMTV/c-neu transgenic animals a positive correlation has previously been shown between the rate of tumor acquisition and the number of pregnancies experienced by the animal.16 Therefore we compared the number of pregnancies achieved by each genotype and found that Casp2−/−/MMTV females had significantly fewer pregnancies before tumor onset (P=0.0238) compared with Casp2+/+/MMTV and Casp2+/−/MMTV animals (Supplementary Figure 1A). Similarly, the average number of pups per litter for Casp2−/−/MMTV females compared with Casp2+/+/MMTV females was also significantly lower (P=<0.0001) (Supplementary Figure 1B). The lower litter size and number of pregnancies observed in the absence of caspase-2 may suggest that caspase-2 has some effect on fertility or fecundity. It is equally possible that Casp2−/−/MMTV mice experience less pregnancies and lower litter sizes as a result of succumbing to more aggressive tumors. However, it is clear from these results that the acceleration of mammary tumor progression observed in multiparous female Casp2−/−MMTV mice is not the result of increased pregnancies or litter size.
Cell morphology of Casp2−/−/MMTVtumors
MMTV/c-neu mice develop luminal tumors and this model clusters with luminal human breast cancer, as opposed to basal-like mammary tumors.17 We stained tumor sections for luminal and basal cells. Tumors from Casp2+/+, Casp2+/− and Casp2−/− MMTV multiparous female mice showed equal staining for cytokeratin 8, a luminal cell marker and no staining for the basal cell marker cytokeratin 5 (Supplementary Figure 2). Thus, tumors from these mice are consistent with a luminal mammary tumor model. Interestingly, we noticed that the nuclei of the luminal cells were greatly enlarged in Casp2−/−/MMTV tumors compared with tumors from wild-type mice.
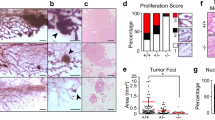
Upon further examination, we observed a number of morphological abnormalities in both Casp2−/−/MMTV and Casp2+/+/MMTV tumors, including karyomegaly (cells display an increase in nuclear size), multinucleated cells, bizarre mitoses (cells with abnormal spindle asters) and necrosis. On the basis of these cellular morphologies, tumors were classified into six different groups that range from less severe to more severe, respectively (Table 1): (1) mild karyomegaly (Figure 2Aa), (2) rarely multinucleated (MN, tumors that rarely exhibited multinucleated cells) (Figure 2Ab), (3) karyomegaly with a slight increase in multinucleated cells (Figure 2Ac and d), (4) karyomegaly with a moderate increase in multinucleated cells (Figure 2Ae), (5) karyomegaly with a large increase in multinucleated cells and cells with bizarre mitoses (Figure 2Af) and (6) necrotic (necrotic tumors were not necessarily more or less severe than other groups, they simply could not be classified).

Tumors from multiparous Casp2−/−/MMTV animals display abnormal morphology. (A) Representative images of the different morphological categories from Casp2+/+/MMTV (a, b) and Casp2−/−/MMTV tumors (c–f) stained with hematoxylin and eosin (H&E) are shown. The morphology of the tumor cells are: mild karyomegaly (a), rarely multinucleated (b), karyomegaly with slight multinucleation (c, d), karyomegaly with moderate multinucleation (e) and karyomegaly with abnormal multinucleated cells and bizarre mitoses (f). Circles indicate areas of abnormal multinucleated cells and arrows indicate bizarre mitotic figures. Scale bars represent 50 microns, images were taken with a 40X objective. (B) Representative images from Casp2+/+/MMTV and Casp2−/−/MMTV animals are shown. Scale bars represent 50 μ, images were taken with a × 40 objective. (C) Increased × 60 magnification of a representative Casp2−/−/MMTV tumor from A (f) is shown. Circles indicate abnormal multinucleated cells and arrows indicate bizarre mitotic figures. Scale bar represents 25 μ
Tumors from multiparous Casp2−/−/MMTV animals were often classified as having karyomegaly with a large increase in multinucleated cells and cells with bizarre mitoses (Figure 2Af, Figure 2B right panel, 2C), while tumors from multiparous Casp2+/+/MMTV tended to only have mild changes in morphology (Table 1A and Figure 2Aa–b, Figure 2B left panel). Casp2+/−/MMTV animals showed a slight increase in tumors characterized as rarely multinucleated and karyomegaly with a slight increase in multinucleated cells compared with wild-type tumors but abnormal karyomegaly and bizarre mitoses were not observed in these tumors. In the nulliparous animals we observed a similar trend, where tumors from Casp2−/−/MMTV animals displayed increased rates of abnormal karyomegaly with multinucleation and bizarre mitoses (Table 1B).
Loss of caspase-2 is associated with cell cycle defects
The abnormal tumor cell morphologies associated with the Casp2−/−/MMTV animals, including bizarre mitoses and multinuclear cells, are consistent with cell cycle defects. These data suggest that tumors lacking Casp2 have accumulated abnormal cells due to the loss of a cell cycle checkpoint control. Consistent with this, tumors from multiparous Casp2−/−/MMTV animals displayed a significantly higher mitotic index (P=0.0498) than tumors from either Casp2+/+/MMTV or Casp2+/−/MMTV multiparous animals (Figure 3a). This difference was not observed in nulliparous animals (Figure 3b). This suggests that Casp2−/−/MMTV tumors proliferate at higher rates than their wild-type counterparts in multiparous animals.

Tumors from multiparous Casp2−/−/MMTV animals display a high mitotic index and abnormal cell cycle regulation. Tumors from multiparous (a) or nulliparous (b) Casp2+/+/MMTV (n=10 multiparous, n=11 nulliparous), Casp2+/−/MMTV (n=11 multiparous, n=11 nulliparous) and Casp2−/−/MMTV (n=20 multiparous, n=8 nulliparous) were examined and the mitotic index was calculated. Mitotic indices for each genotype are shown. Error bars represent the S.E.M. P-values were determined by one-way ANOVA. (c–f) Casp2+/+ and Casp2−/− MEF were treated left untreated (c), or treated with 1 μ M 5-Fluorouracil (d), 1 ng/ml nocodazole (e), or cultured in the absence of serum (f) for 16 h, at which point the media was replaced with fresh media and cells were cultured for an additional 24 h. Cells were harvested and the cell cycle was analyzed by propidium iodide staining. The percentage of cells in G1, S or G2/M phase for each cell line is shown. Error bars represent the S.D. of at least three separate experiments
To further examine the effect of caspase-2 on cell cycle progression in transformed cells, we examined cell cycle profiles of E1A/Ras-transformed MEF. In cycling cells, we observed a consistently higher number of Casp2−/− MEF in the G2/M phase of the cell cycle compared with wild-type cells (Figure 3c). This profile is consistent with cells that have higher proliferation rates, suggesting that more cells are in the process of entering mitosis. We synchronized the cells by arresting cells with 5-FU (Figure 3d), nocodazole (Figure 3e) or serum withdrawal (Figure 3f), and then released cells into cycle again. In each case, 24 h following synchronization, we observed a similar increase in the proportion of cells in G2/M in the absence of caspase-2, further suggesting that the loss of caspase-2 results in increased proliferation of transformed cells.
Loss of caspase-2 increases genomic instability
The observed increase in cell proliferation and accumulation of cell cycle defects in the absence of caspase-2 suggest that tumor cells lacking caspase-2 may be more prone to genomic instability. To investigate this, we performed in vitro PALA (N-(phosphonoacetyl)-L-aspartate) assays in E1A/Ras-transformed Casp2+/+ and Casp2−/− MEF. PALA is a pyrimidine synthesis inhibitor that induces G1-arrest.18, 19 Genomically unstable cells become resistant to PALA by amplifying Cad (carbamyl phosphate synthetase/aspartate transcarbamylase/dihydro-orotase20), which encodes an essential enzyme in the synthesis of UMP.21
In initial experiments, aimed at determining the sensitivity of E1A/Ras-transformed Casp2+/+ and Casp2−/− MEF, we observed that cells lacking caspase-2 were more resistant to PALA treatment (LD50=∼50 μ M) than wild-type cells (LD50=∼10 μ M, Supplementary Figure 3A). We confirmed these results in an independently-derived set of litter-matched Casp2+/+ and Casp2−/− MEF (Supplementary Figure 3B). Knockdown of caspase-2 in MCF7 cells using short hairpin RNA (shRNA) (Figure 5c), similarly conferred increased resistance to PALA treatment and knockdown cells had higher LD50s compared with cells transduced with the control shRNA sequence (>100 μ M versus 80 μ M, Supplementary Figure 3C).
When Casp2+/+and Casp2−/−MEF were challenged for 2 weeks with doses equivalent to three times and five times their respective LD50 of PALA, we observed that Casp2−/− MEF were profoundly resistant to PALA (Figure 4a). Accordingly, we also observed a greater increase in genomic Cad amplification in Casp2−/− MEF compared with Casp2+/+ MEF following prolonged exposure to PALA (Figure 4b). Together these results suggest that caspase-2 contributes to maintaining genomic instability.

Loss of caspase-2 promotes PALA resistance. (a) E1A/Ras-transformed Casp2+/+ MEF and Casp2−/− MEF from littermate embryos were treated with 3 × and 5 × their respective LD50 doses of PALA (see Supplementary Figure 3). Two weeks after treatment, cells were stained with Giemsa and imaged. (b) Genomic DNA from the cells in (a) was isolated and levels of genomic Cad in nanograms (ng) were analyzed by quantitative real-time PCR and normalized to genomic levels of 18s in ng. Error bars represent the S.D. of triplicate determinations. Results are representative of three independent experiments
Loss of caspase-2 is associated with an impaired p53 response to PALA
Loss of p53 is known to enhance genomic instability,18, 19 therefore we sequenced the p53 gene in Casp2+/+ or Casp2−/− MEF to rule out the possibility that p53 had been differentially lost or spontaneously mutated, accounting for the differences in genomic instability we observed. The p53 sequence was identical in both cell lines and did not harbor any mutations (Supplementary Figure 4). We also measured p53 protein levels by western blot and p53 was readily detected in untreated Casp2−/− MEF (Figure 5a, Supplementary Figure 5A). Together these results indicate that Casp2−/− MEF maintain wild-type p53 that is expressed and thus the increased genomic instability observed is unlikely to be due to inactivating mutations in p53 in the knockout cell line.

Caspase-2-deficient cells have an impaired p53 response to PALA and etoposide. (a) Casp2+/+ and Casp2−/− MEF were harvested at the indicated times after treatment with 100 μ M PALA and analyzed by immunoblot for p53, p21, caspase-2 and actin as a loading control. (b) Casp2+/+ and Casp2−/− MEF were treated as in (a). cDNA was analyzed by quantitative real-time PCR analysis for Mdm2, p21 and Pidd. Absolute mRNA values were determined from a standard curve and normalized to 18s. Error bars represent the S.D. of triplicate determinations. (c) MCF7 cells were stably transfected with non-targeting shRNA (shControl) or two different shRNAs targeting CASP2 (shCASP2 no. 1 and shCASP2 no. 2). Knockdown of caspase-2 in MCF7 cells was detected by immunoblot. (d) MCF7 cells and caspase-2 knockdown MCF7 cells were treated as in (a) and absolute p21 mRNA values were determined as in (b). Error bars represent the S.D. of triplicate determinations. (e) Casp2+/+ and Casp2−/− MEF were treated with 10 μ M etoposide and analyzed for Mdm2 and p21 expression as in (b). Error bars represent the S.D. of triplicate determinations
Interestingly, the level of p53 protein was actually increased in Casp2−/− MEF compared with the wild-type cells (Figure 5a, Supplementary Figure 5A). In addition, while p53 was stabilized after treatment with PALA in wild-type cells, further stabilization of p53 in Casp2−/− cells in response to PALA was not detected. We also noted slightly increased basal protein levels of the p53 target p21 in the absence of caspase-2 compared with wild-type cells by western blot (Figure 5a). To explore this further, we used quantitative real-time PCR to measure basal mRNA levels of p53 and p21 in Casp2+/+and Casp2−/− MEF (Supplementary Figure 5B). There was little change in p53 expression levels in the absence of caspase-2 compared with wild-type cells. However, p21 basal levels were slightly increased in the absence of caspase-2. This may suggest that the increased protein levels of p53 reflect increased protein stability rather than increased expression. Interestingly, treatment with PALA failed to further induce p21 protein levels in Casp2−/− MEF while induction of p21 was apparent in the wild-type cells (Figure 5a). These results suggest that while p53 may be constitutively active in the absence of caspase-2, the p53 response to PALA may be impaired.
To further test the effect of caspase-2 on the p53 response to PALA, we used quantitative real-time PCR to measure the induction of p53 target genes. PALA induced robust induction of p21, Mdm2 and Pidd in a p53-dependent manner (Figure 5b and Supplementary Figure 5C). However, in Casp2−/− MEF, induction of p21, Mdm2 and Pidd induced by PALA was decreased. We also stably knocked down caspase-2 in MCF7 cells using two different shRNA sequences and observed that induction of p21 in response to PALA was similarly reduced when caspase-2 was deficient (Figure 5d). These results suggest that the p53 response to PALA is somewhat impaired in the absence of caspase-2.
Finally, we treated cells with the DNA damaging agent etoposide, which engages the p53 pathway. Similar to what we observed in response to PALA treatment, etoposide induced Mdm2 and p21 mRNA in MEF in a p53-dependent manner (Supplementary Figure 5D). In caspase-2-deficient MEF, treatment with etoposide led to an overall reduction of Mdm2 and p21 mRNA to levels lower than in wild-type MEF. These results suggest that etoposide-induced expression of p53 target genes is also somewhat blunted in the absence of caspase-2 (Figure 5e).
Discussion
Caspase-2 has previously been implicated as a tumor suppressor in Eμ-myc-driven lymphomagenesis.2, 22 We show here that caspase-2 can also suppress epithelial tumors as we demonstrate a previously unknown role for caspase-2 as a tumor suppressor in breast cancer.
We report that multiparous mice lacking caspase-2 acquired tumors at an increased rate resulting in impaired survival. MMTV/neu mice used in this study were on an FVB/N background. FBV MMTV mice develop mammary tumors between 7 and 12 months of age. Mice on a C57BL/6 background are quite resistant to MMTV/c-neu-driven tumors and tumor onset in FVB × C57BL/6 (F1) MMTV/Neu is delayed to over 18 months.23, 24 Caspase-2 mice used here were on a mixed 129/C57BL/6 background. It has been previously shown that 129/C57BL/6 × FVB MMTV/Neu (F1) nulliparous mice developed mammary tumors by 11 months, consistent with the tumor latency on a pure FVB background, suggesting that the mixed 129/C57BL/6 × FVB background does not impede or accelerate progression of tumorigenesis.25 The median survival of MMTV/Casp2+/+ females was 321 days and 241 days for nulliparous and multiparous females, respectively. This is very similar to previously published median survival times of ∼300 days and 250 days, respectively for MMTV/Neu mice bred on a pure FVB background.16 Therefore, the acceleration of mammary tumorigenesis observed in the absence of caspase-2 cannot be explained by background genetic effects.
Functional differentiation of the mammary gland does not occur until pregnancy and lactation, therefore, in multiparous female mice the mammary gland experiences rounds of proliferation followed by cell death during involution and lactation that are not experienced by nulliparous animals contributing to decelerated tumorigenesis in nulliparous MMTV animals.26 Caspase-2-deficient nulliparous mice showed no alteration in rate of tumor acquisition or mitotic index but the morphological abnormalities were present. This suggests that caspase-2 prevents cellular aberrations, such as multinucleation and bizarre mitoses in addition to attenuating progression of these damaged cells when proliferation rates are high.
Loss of one copy of caspase-2 was sufficient to partly accelerate mammary tumorigenesis and impair survival in multiparous animals but no increase in mitotic index was observed and the cellular abnormalities were not as severe. Loss of both copies of caspase-2 resulted in a far more pronounced and significant survival impairment. This suggests that complete loss of caspase-2 is associated with more aggressive forms of disease as abnormal mitotic figures, such as those that we observed with high frequency in caspase-2-deficient tumors, usually correlate with aggressive tumor growth and poor prognosis. For example, tripolar mitoses (Figure 2Ad) are considered to be definitive evidence of malignancy.27 The small survival impairment associated with loss of one copy of caspase-2 may be due to defects that are below the threshold of detection in the assays used here or the slight increase in the less severe abnormalities, such as multinucleation were sufficient to promote the partial acceleration of tumorigenesis observed in heterozgote animals.
The higher mitotic index and cellular abnormalities consistent with cell cycle defects observed in caspase-2-deficient mammary tumors are consistent with evidence suggesting that caspase-2 has a role in regulating cell cycle events.2, 9, 10 In addition, we observed that caspase-2-deficient MEF tended to accumulate in the G2/M phase of the cell cycle. This is in line with observations by Manzl et al.,22 who noted a similar trend in caspase-2-deficient Eμ-Myc lymphoma cells suggesting that caspase-2 promotes correct G2/M transition. Therefore, caspase-2 may function as a tumor suppressor, in part, through correct regulation of cell cycle progression.
Abnormal mitoses usually result from a failure of the spindle assembly checkpoint and often result in genomic instability and aneuploidy. We observe resistance to PALA and amplification of genomic Cad in the absence of caspase-2 suggesting that caspase-2 prevents genomic instability in transformed cells. Consistent with our results, Dorstyn et al.9 recently showed that caspase-2-deficient MEF and lymphoma cells exhibit aneuploidy, again suggesting that caspase-2 prevents acquisition of chromosomal aberrations in response to transformation.
Loss of p53 often results in genomic instability as measured by resistance to PALA and gene amplification.18, 19 PALA induces chromosomal breaks by inhibiting pyrimidine nucleotide synthesis and often results in p53-dependent cell cycle arrest at G1.18, 19 It has been shown that p53 is activated by metabolic depletion induced by PALA before any DNA damage occurs and thus p53 may be activated at multiple points in this pathway.28
We observed that, while p53 expression and sequence was intact in caspase-2-deficient cells, the expression of p53-dependent transcriptional targets, including Mdm2, p21 and Pidd in response to PALA was reduced. This was despite the fact that p53 appears to be inherently more stable in caspase-2-deficient cells. The increased stability of p53 and increased basal levels of p21 in the absence of caspase-2 may be a consequence of increased proliferation rates of caspase-2-deficient cells, which has been observed here and reported by others previously.2, 22 It has been proposed that caspase-2 may enhance p53 function by cleaving MDM2 to a fragment that binds p53 but does not degrade it,29 resulting in a feed-forward loop where stabilization of p53 induces PIDD upregulation and increased caspase-2 activity.29 Our observations may provide evidence in favor of this model, where caspase-2 activity is not required for the p53 response to PALA but does enhance it. We also observed an additional interesting parallel between caspase-2 and p53. p53 knockout mice have been shown to have defects in fertility and fecundity.30 Our observations that Casp2−/−/MMTV mice have significantly lower numbers of pregnancies and litter sizes may reflect an additional physiological role for caspase-2 in the p53 pathway.
DNA damage can trigger a p53-independent ATM/ATR pathway that has been shown to induce caspase-2-dependent apoptosis when Chk1 is suppressed.7 Therefore it is possible that there is a p53-independent component to the role of caspase-2 in maintaining genomic stability that is impacted by the Chk1-suppressed pathway or other p53-indepenent pathways to caspase-2 activation.
Our findings support the idea that deletion, epigenetic silencing or transcriptional and/or translational inhibition of caspase-2 activity may be involved in the progression of breast cancer and other epithelial cancers. According to the Cancer Genome Project, 10/45 breast tumors examined displayed reduction of copy number in the chromosomal region encompassing CASP2 as measured by loss of heterozygosity (LOH) (http://www.sanger.ac.uk/cgi-bin/genetics/CGP/conan/search.cgi). Thus, there may be haplo-insufficiency of caspase-2 in certain human mammary tumors. Microarray analysis of 120 gastric tumors revealed that caspase-2 was only expressed in 35% of tumor samples despite high expression in normal gastric tissue.31 Downregulation of caspase-2 expression has been reported in renal cell carcinoma,32, 33 and missense mutations in CASP2 have been described in lung and ovarian tumors.34 The promoter region of CASP2 contains a CpG island35 and may be subject to methylation-mediated epigenetic silencing in lung adenocarcinomas.36
In summary, our in vitro data, combined with in vivo data examining morphology and mitotic index of Casp2−/−/MMTV tumors, supports a tumor suppressive function for caspase-2 as a regulator of the cell cycle and genomic instability. We therefore conclude that caspase-2 is a tumor suppressor in epithelial cancers such as breast cancer, and may be an important genetic marker for aggressive disease and poor prognosis.
Materials and Methods
Cell lines and reagents
Casp2+/+ and Casp2−/− MEF were generated from day 13.5 embryos as follows. Briefly, the head and the red organs were removed and then the torso was minced and dispersed in DMEM containing 10% FCS, 2 mM L-glutamine, 50 I.U. Penicillin/50 μg/ml Streptomycin, 1 mM sodium pyruvate, 1 × non-essential amino acids and 55 μ M β-mercaptoethanol. Early-passage MEF were simultaneously transduced with frozen supernatants of the retroviral expression vectors pBabePuro.H-ras (G12V) and pWZLH.E1A (provided by SW Lowe and G Hannon). After 48 h, the cells were harvested by mild trypsinization, seeded at 1 × 105 cells/well in six-well plates and cultured for 10 days in media containing 0.5 μg/ml of puromycin and 40 μg/ml of hygromycin for selection of the transduced viruses. Genomic DNA was extracted from the embryo heads and used for genotyping analysis. Wild-type MEF lines were derived from the same litter. p53+/+ and p53−/− litter-matched E1A/Ras-transformed MEF were a gift from Gerard Zambetti (St. Jude Children’s Hospital, Memphis, TN, USA) and were cultured in the same media as Casp2+/+ and Casp2−/− MEF. MCF7 breast carcinoma cells were cultured in DMEM containing 10% FCS, 2 mM L-glutamine and 50 I.U. Penicillin/50 μg/ml Streptomycin. All media reagents were purchased from Invitrogen (Carlsbad, CA, USA).
Generation of Casp2+/+/MMTV, Casp2+/−/MMTV and Casp2−/−/MMTV mice
Mice with a homozygous deletion of Casp2 on a mixed 129/B6 background (described previously in Bergeron et al.37) were mated to Casp2+/+ male mice expressing one allele of MMTV/c-neu on an FVB background (purchased from Jackson Laboratory, Bar Harbor, ME, USA). Resulting Casp2+/−/MMTV male mice were then mated to Casp2+/−/MMTV-negative female mice, generating Casp2+/+/MMTV, Casp2+/−/MMTV and Casp2−/−/MMTV mice. Females from this cross were then mated (multiparous) or housed as virgins (nulliparous). Animals were examined every other day for the appearance of tumors. When animals became moribund due to tumor size, they were submitted to the St. Jude Veterinary Pathology Core facility for routine necropsy and histology. Genotyping was confirmed by tail snip PCR as described previously.37 The St Jude Institutional Animal Care and Use Committee approved all procedures in accordance with the Guide for the Care and Use of Animals. The data shown represents all animals in the study. We also examined the disease progression in individual litters to further rule out effects of genetic background and observed the same trend of accelerated tumorigenesis in Casp2−/−/MMTV animals (data not shown).
MMTV tumor studies
When animals became moribund (e.g. tumor burden impaired movement or otherwise recommended euthanization), mice were euthanized via inhalation with 2% isoflurane. Mammary tumors were collected and fixed in 10% neutral buffered formalin. Specimens were processed routinely, paraffin embedded, cut at 4 μ, stained with hematoxylin and eosin, and analyzed by light microcopy. Images were acquired using an Olympus BX41 light microscope with a Spot Insight Color Mosaic camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA) and Spot imaging software (Diagnostic Instruments Inc.,).
Tumor morphology and mitotic index
Tumors were examined by a veterinary pathologist for qualitative assessment of morphologic features. Luminal and basal cells were assessed by staining with cytokeratin 8 and cytokeratin 5 antibodies, respectively. Briefly, slides of 4 μ tumor sections were cut from formalin-fixed paraffin-embedded tissues. All assay steps, including deparaffinization, rehydration and epitope retrieval, were performed on the Bond Max with Bond wash buffer (cat no. AR9590, Leica, Bannockburn, IL, USA) and rinses between steps. Heat-induced epitope retrieval was performed by heating slides in ER1 (cat no. AR9961, Leica) for 20 min. The Refine (cat no. DS9800, Leica) detection system was used. Slides were incubated sequentially with hydrogen peroxide (5 min), primary antibody (15 min), postprimary (8 min), anti-rabbit horse radish peroxidase conjugated polymer (8 min), 3,3′ diaminobenzidine (10 min) and hematoxylin (5 min). The primary antibodies were obtained from Thermo Scientific (Kalamazoo, MI, USA) (Cytokeratin 5, cat no. RM-2106-R7 and Cytokeratin 8, cat no. RM-2107-R7, Thermo Scientific) and used as provided at a concentration of 0.05 mol/l. Mitotic index was determined by counting the number of mitotic figures in × 10–400 fields in a representative tumor from the cervical, thoracic and inguinal mammary glands (when present), and averaged across these areas.
Cell cycle analysis
Casp2+/+ and Casp2−/− MEF were plated 1 day before treatment with the indicated drugs. Cells were treated with 1 μm 5-Fluorouracil (Enzo Life Sciences, Farmingdale, NY, USA), 1 ng/ml nocodazole (Acros Organics, Fair Lawn, NJ, USA), or cultured in media depleted of serum. After 16 h, the media was replaced with fresh complete media and cells were cultured for an additional 24 h. For cell cycle analysis, cells were harvested by trypsinization, washed once in cold PBS, and resuspended in 1 ml of cold PBS. Cell suspensions were added dropwise to 1 ml of cold 100% ethanol and incubated overnight at 4 °C. After overnight incubation, cells were centrifuged at 1500 × g at 4 °C for 10 min, washed once with cold PBS, and centrifuged again at 1500 × g at 4 °C for 10 min. Cell pellets were then resuspended in PBS containing 0.1% Triton-X 100, 20 μg/ml PI (Sigma, St. Louis, MO, USA) and 2 mg/ml DNase-free RNaseA (Roche, Indianapolis, IN, USA) and incubated for 15 min at 37 °C. Cells were then analyzed by flow cytometry using standard cell cycle analysis protocols.
Knockdown of CASP2
shRNA GIPZ lentiviral constructs targeting human CASP2 (NM_032982) were obtained from Baylor College of Medicine Cell-Based Assay Screening Service (C-BASS, Houston, TX, USA). The sequences of the shRNAs used were: shCASP2 no. 1 – 5′-TGCAGAATTTTGCACAGTT-3′; shCASP2 no. 2 – 5′-CGGTGCAAGGAGATGTCTG-3′; and shControl (non-silencing) –5′-ATCTCGCTTGGGCGAGAGTAAG-3′. Lentiviral particles were generated using the Trans-Lentiviral Packaging System (Thermo Scientific) according to the manufacturer’s instructions. The lentiviral particles were used to transduce MCF7 cells. Forty-eight hours after transduction, MCF7 cells were selected in 1 μg/ml puromycin. Knockdown of caspase-2 in the resulting puromycin-resistant cells was verified by western blot analysis (see Figure 4).
PALA assay
The LD50 of PALA for the indicated cell lines was determined as follows: cells were seeded in 96-well plates at a density of 1 × 104 cells/ml. The following day, cells were incubated for 3 days with the indicated doses of PALA (Drug Synthesis and Chemistry Branch, National Cancer Institute) in α-MEM containing 50 I.U. Penicillin/50 μg/ml Streptomycin (Invitrogen) and 10% dialyzed fetal bovine serum (Invitrogen). Cell viability was determined by XTT assay, performed according to manufacturer’s instructions (Cell Signaling Technologies, Beverly, MA, USA). For long-term PALA challenge, cells were plated in triplicate at a density of 1 × 104 cells/ml in 10 cm2 tissue culture dishes. The following day, cells were treated with three times and five times their respective LD50 dose of PALA. Media was changed every 3-4 days for 2 weeks. Plates were stained with Giemsa, and genomic DNA was harvested using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA).
Western blotting
Cells were treated as indicated below. Cells were harvested by trypsinization at the indicated times and lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 5 mM EDTA, 1% NP-40 and 1% Triton-X 100 containing 1 × protease inhibitors (Complete Mini Protease Inhibitor Cocktail, Roche), 10 mM sodium fluoride and 1 mM sodium orthovanadate (Sigma). Protein concentration was determined by BCA assay (Pierce, Rockford, IL, USA). A total of 100–150 μg protein was separated on Criterion-XT gels (Bio-Rad, Berkley, CA, USA) and transferred onto Hybond XL membrane (GE Healthcare Life Sciences, Pittsburg, PA, USA), and probed with the indicated antibodies. Antibodies used were: anti-caspase-2 (Clone 11B4, EMD Millipore, Billerica, MA, USA), anti-p53 (Clone IC12, Cell Signaling Technologies), anti-p21 (Clone SXM30, BD Biosciences, San Jose, CA, USA) and anti-actin (Clone C4, MP Biomedicals, Santa Ana, CA, USA). Secondary anti-mouse-HRP and anti-rat-HRP were purchased from GE Healthcare Life Sciences. Antibody-HRP complexes were detected using West Pico and West Dura Substrate (Pierce).
Quantitative real-time PCR analysis
For analysis of gene expression following treatment with PALA or etoposide (Sigma), or genomic Cad following long-term treatment with PALA, cells were harvested by trypsinization at the indicated times, and total RNA was prepared using the RNeasy Kit (Qiagen). Total RNA was treated with amplification-grade DNase I and reverse-transcribed using random hexamer priming and MMLV-reverse transcriptase (Invitrogen) Samples were amplified in triplicate using iQ SYBR Green Supermix (Bio-Rad) and the MyiQ real-time detection system. Melt curve analysis was performed to ensure a single product species. Relative starting quantities were calculated by comparison to a common standard curve generated with a dilution series of unrelated murine cDNA or genomic DNA (for analysis of gene expression or genomic Cad levels, respectively, in MEF) or cDNA from MB231 breast carcinoma cells (for MCF7 cells) that was included in each run. For control 18s was used. Real-time primers (ordered from Sigma Genosys, The Woodlands, TX, USA) were: murine Mdm2 – 5′-CAGAGACGCCCTCGCATC-3′ (sense) and 5′-CTGAATCCTGATCCAGGCAATC-3′ (antisense); murine p21 – 5′-CCGTTGTCTCTTCGGTCCC-3′ (sense) and 5′-CATGAGCGCATCGCAATC-3′ (antisense); murine Pidd – 5′-GGGAACCAGTTGAACTTGGAC (sense) and 5′-CTGAGCCGCAAAAACTCCAC-3′ (antisense); murine 18s – 5′-ATGGTAGTCGCCGTGCCTAC-3′ (sense) and 5′-CCGGAATCGAACCCTGATT-3′ (antisense); human p21 – 5′-AGCGACCTTCCTCATCCA-3′ (sense) and 5′-ATAGCCTCTACTGCCACCAT-3′ (antisense); and human 18s – 5′-GAGGGAGCCTGAGAAACGG-3′ (sense) and 5′-GTCGGGAGTGGGTAATTTGC-3′ (antisense); murine Cad – 5′-AAG CTC AGA TCC TAG TGC TAA CG-3′ (sense) and 5′-CCGTAGTTGCCGATGAGAGG-3′ (antisense).
Genomic p53 sequencing
Genomic DNA was isolated from Casp2+/+ and Casp2−/− MEF using the DNeasy Blood and Tissue Kit (Qiagen). Exons 3–10 (corresponding to the coding sequence of murine p53) were amplified by standard PCR. PCR products were gel-purified before cloning with the TOPO-TA Cloning Kit for Sequencing (Invitrogen) as per the manufacturer’s instructions. The sequences of the primers were as follows: exons 3 and 4, 5′-TGGATGTCCCACCTTCTTT-3′ (sense) and 5′-GAACCCTCAGGACTGTGTTCTCAGC-3′ (antisense); exon 5, 5′- GAAGATATCCTGGTAAGGCCCAGAGCA-3′ (sense) and 5′-AGCGGCATTGAAAGGTCACACGAAAG-3′ (antisense); exons 6 and 7, 5′-AGTTCCCCACCTTGACACCT-3′ (sense) and 5′-GTCAACTGTCTCTAAGACGCA-3′ (antisense); exon 8, 5′-TGTAGTGAGGTAGGGAGCGAC-3′ (sense) and 5′- CTGGGGAAGAAACAGGCTAA-3′ (antisense); exons 9 and 10, 5′-GTGCTGGTCCTTTTCTTGT-3′ (sense) and 5′-GCAATAATGGGTACAGCTTG-3′ (antisense).
Abbreviations
- 5-FU:
-
5-Fluorouracil
- ALL:
-
acute lymphoblastic leukemia
- AML:
-
acute myeloid leukemia
- ATM:
-
ataxia telangiectasia mutated
- ATR:
-
ataxia telangiectasia and Rad3-related protein
- CAD:
-
carbamyl phosphate synthetase/aspartate transcarbamylase/dihydro-orotase
- Chk1:
-
checkpoint kinase 1
- MDM2:
-
mammalian double minute 2
- MEF:
-
mouse embryonic fibroblasts
- MMTV:
-
mouse mammary tumor virus
- MN:
-
multinucleated
- PALA:
-
N-(phosphonoacetyl)-L-aspartate
- PIDD:
-
p53-induced protein with a death domain
- shRNA:
-
short hairpin RNA
- T-ALL:
-
T cell acute lymphoblastic leukemia
References
Bouchier-Hayes L, Green DR . Caspase-2: the orphan caspase. Cell Death Differ 2012; 19: 51–57.
Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S . A tumor suppressor function for caspase-2. Proc Natl Acad Sci USA 2009; 106: 5336–5341.
Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR . In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol 2006; 8: 72–77.
Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S . Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 2008; 27: 3393–3404.
Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML . Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci 2000; 20: 1386–1392.
Stefanis L, Troy CM, Qi H, Shelanski ML, Greene LA . Caspase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity. J Neurosci 1998; 18: 9204–9215.
Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008; 133: 864–877.
Nutt LK, Buchakjian MR, Gan E, Darbandi R, Yoon SY, Wu JQ et al. Metabolic control of oocyte apoptosis mediated by 14-3-3zeta-regulated dephosphorylation of caspase-2. Dev Cell 2009; 16: 856–866.
Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ 2012; 19: 1288–1298.
Andersen JL, Johnson CE, Freel CD, Parrish AB, Day JL, Buchakjian MR et al. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J 2009; 28: 3216–3227.
Holleman A, den Boer ML, Kazemier KM, Beverloo HB, von Bergh AR, Janka-Schaub GE et al. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood 2005; 106: 1817–1823.
Estrov Z, Thall PF, Talpaz M, Estey EH, Kantarjian HM, Andreeff M et al. Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia. Blood 1998; 92: 3090–3097.
Faderl S, Estrov Z . Detection of residual disease in childhood acute lymphoblastic leukemia. N Engl J Med 1999; 340: 152–153 author reply 153–154.
Faderl S, Estrov Z . The clinical significance of caspase regulation in acute leukemia. Leuk Lymphoma 2001; 40: 471–481.
Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P . Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988; 54: 105–115.
Anisimov VN, Popovich IG, Alimova IN, Zabezhinski MA, Semenchenko AV, Yashin AI . Number of pregnancies and ovariectomy modify mammary carcinoma development in transgenic HER-2/neu female mice. Cancer Lett 2003; 193: 49–55.
Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 2007; 8: R76.
Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD . Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 1992; 70: 923–935.
Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM . Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell 1992; 70: 937–948.
Smith KA, Chernova OB, Groves RP, Stark MB, Martinez JL, Davidson JN et al. Multiple mechanisms of N-phosphonacetyl-L-aspartate resistance in human cell lines: carbamyl-P synthetase/aspartate transcarbamylase/dihydro-orotase gene amplification is frequent only when chromosome 2 is rearranged. Proc Natl Acad Sci USA 1997; 94: 1816–1821.
Wahl GM, Padgett RA, Stark GR . Gene amplification causes overproduction of the first three enzymes of UMP synthesis in N-(phosphonacetyl)-L-aspartate-resistant hamster cells. J Biol Chem 1979; 254: 8679–8689.
Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ 2012; 19: 1722–1732.
Rowse GJ, Ritland SR, Gendler SJ . Genetic modulation of neu proto-oncogene-induced mammary tumorigenesis. Cancer Res 1998; 58: 2675–2679.
Taneja P, Frazier DP, Kendig RD, Maglic D, Sugiyama T, Kai F et al. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn 2009; 9: 423–440.
Maroulakou IG, Oemler W, Naber SP, Tsichlis PN . Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res 2007; 67: 167–177.
Strange R, Li F, Saurer S, Burkhardt A, Friis RR . Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development 1992; 115: 49–58.
Gisselsson D, Jin Y, Lindgren D, Persson J, Gisselsson L, Hanks S et al. Generation of trisomies in cancer cells by multipolar mitosis and incomplete cytokinesis. Proc Natl Acad Sci USA 2010; 107: 20489–20493.
Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM . A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev 1996; 10: 934–947.
Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell 2011; 43: 57–71.
Hu W, Feng Z, Teresky AK, Levine AJ . p53 regulates maternal reproduction through LIF. Nature 2007; 450: 721–724.
Yoo NJ, Lee JW, Kim YJ, Soung YH, Kim SY, Nam SW et al. Loss of caspase-2, -6 and -7 expression in gastric cancers. Acta Pathol Microbiol Scand 2004; 112: 330–335.
Heikaus S, Pejin I, Gabbert HE, Ramp U, Mahotka C . PIDDosome expression and the role of caspase-2 activation for chemotherapy-induced apoptosis in RCCs. Cell Oncol 2010; 32: 29–42.
Jones TD, Eble JN, Wang M, MacLennan GT, Delahunt B, Brunelli M et al. Molecular genetic evidence for the independent origin of multifocal papillary tumors in patients with papillary renal cell carcinomas. Clin Cancer Res 2005; 11: 7226–7233.
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010; 466: 869–873.
Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39: 457–466.
Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S et al. Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res 2009; 69: 6315–6321.
Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 1998; 12: 1304–1314.
Acknowledgements
This work was supported by the National Institutes of Health AI47891 (DRG) and 5F32CA1360912 (MJP). We thank An Lu for technical help and Jonathan Flanagan for helpful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Parsons, M., McCormick, L., Janke, L. et al. Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice. Cell Death Differ 20, 1174–1182 (2013). https://doi.org/10.1038/cdd.2013.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.38
Keywords
This article is cited by
-
A Review on Caspases: Key Regulators of Biological Activities and Apoptosis
Molecular Neurobiology (2023)
-
The caspase-2 substrate p54nrb exhibits a multifaceted role in tumor cell death susceptibility via gene regulatory functions
Cell Death & Disease (2022)
-
Caspase-2 regulates S-phase cell cycle events to protect from DNA damage accumulation independent of apoptosis
Oncogene (2022)
-
Phosphorylation by Aurora B kinase regulates caspase-2 activity and function
Cell Death & Differentiation (2021)
-
The p53-caspase-2 axis in the cell cycle and DNA damage response
Experimental & Molecular Medicine (2021)
