
Abstract
Heavy alcohol consumption provokes an array of degenerative pathologies but the signals that couple alcohol exposure to regulated forms of cell death are poorly understood. Using Drosophila as a model, we genetically establish that the severity of ethanol challenge dictates the type of death that occurs. In contrast to responses seen under acute exposure, cytotoxic responses to milder challenges required gene encoding components of the apoptosome, Dronc and Dark. We conducted a genome-wide RNAi screen to capture targets that specifically mediate ethanol-induced cell death. One effector, Drat, encodes a novel protein that contains an ADH domain but lacks essential residues in the catalytic site. In cultured cells and neurons in vivo, depletion of Drat conferred protection from alcohol-induced apoptosis. Adults mutated for Drat showed both improved survival and enhanced propensities toward sedation after alcohol challenge. Together, these findings highlight novel effectors that support regulated cell death incited by alcohol stress in vitro and in vivo.
Similar content being viewed by others


Main
Alcohol abuse is a worldwide problem and knowledge of its toxic properties can be traced to the beginning of recorded medical history.1 Excessive ethanol consumption is associated with dementia, liver injury, hypertension, compromised immunity, cardiomyopathy, enhanced cancer risk and neurobehavioral problems. Exposure to alcohol in utero leads to diminished brain size and lifelong neurobehavioral disturbances commonly referred to as fetal alcohol syndrome (FAS).2
Evidence that ethanol is directly injurious to cells and tissues is uncontested but the precise mechanistic basis for cytotoxicity is ill defined. A confounding problem traces to the fact that alcohol stress provokes many histological changes but it is typically not clear whether these alterations are the cause or the consequence of a pending cell death response. Furthermore, it is equally unclear whether ethanol or downstream metabolites are responsible. High alcohol concentrations (>5%) cause immediate destruction, essentially solubilizing cell membranes and dramatically changing tertiary protein structure.3 At lower concentrations, regulated forms of cell death occur and mediators considered important for ethanol-induced cytotoxicity include both oxidative and on-oxidative metabolites (reviewed in Dey and Cederbaum4 and Hoek and Pastorino5). Non-oxidative metabolism of ethanol results in the production of ethanol-containing phospholipids and fatty acid ethyl esters. These metabolites may directly perturb the physical properties of cell membranes and further contribute to cell death by interfering with synthesis of DNA, protein and normal signal transduction events. In addition, oxidative metabolism of ethanol produces acetaldehyde and diverse reactive oxygen species, resulting in increased oxidative stress. Cytosolic alcohol dehydrogenase (ADH) appears to be the major pathway for ethanol oxidation under normal conditions.
Other cellular responses provoked by ethanol treatment include changes in calcium homeostasis, modified differentiation, inhibition of proliferation, induced cytokine production and altered mitochondrial permeability.3, 6, 7 Possibly as a result of these diverse and pleiotropic effects, no single mechanism has been identified to fully account for ethanol-induced cytotoxicity. An emerging consensus is that cellular responses are qualitatively affected by the level and frequency of exposure. Acutely high levels of ethanol cause direct and irreversible damage to cells, sometimes evoking necrotic forms of cell death, whereas subacute exposure to this stressor may provoke a variety of cellular responses, many of which trace to mitochondrial dysfunction and/or programmed pathways of cell death.7
Apoptotic cell death is commonly associated with alcohol-induced pathologies. Over the past decade, studies from several experimental models and human biopsies have tightly correlated induced apoptosis with ethanol-induced damage in a variety of tissues including liver, buccal mucosa, salivary gland, gastric mucosa, brain, thymus, spleen and gut-associated lymphoid tissue.8, 9, 10, 11 Ethanol exposure during gastrulation and early neurulation induces apoptosis within certain embryonic cell populations, leading to mental retardation and neurobehavioral disorders, as well as FAS.12 In the liver, alcohol-induced apoptosis is an essential feature contributing to chronic pathologies of this organ, such as alcoholic hepatitis and steatosis.13, 14, 15, 16 Hepatocyte apoptosis may define at least a part of the ‘inciting event’ that instigates stellate cell activation and fibrogenesis during cirrhosis.17 Although certain genes (e.g. Bax, Bcl-XL, phospholipase C and p53) have been implicated as effectors of alcohol toxicity, the signaling pathways that couple alcohol stress to apoptosis are not known.10, 18, 19, 20
Drosophila is a well-established model system for alcohol research.21, 22 Flies and humans share common enzymatic pathways for metabolizing ethanol (e.g. Adh, acetaldehyde dehydrogenase (Aldh)), and intoxication behaviors associated with altered neurophysiologies are starkly similar.21, 22, 23 Drosophila is also a premiere system for examining gene function during programmed and stress-induced cell death (reviewed in Hay and Guo24). As in humans, intrinsic pathways of apoptosis are controlled by an apoptosome composed of Dronc (an ortholog of Caspase-9) and its adapter, Dark (an ortholog of Apaf-1). Using an ex vivo culture system to test blood cells (hemocytes) isolated from mutant animals, we previously showed that both Dronc and Dark were required for alcohol-induced cell killing.25 Therefore, in this system, alcohol-induced toxicity clearly proceeds through an apoptosome-dependent pathway.
To identify signaling events associated with alcohol stress and further understand how these are integrated to provoke regulated cell death, we developed a model of ethanol-induced apoptosis in cultured fly cells. As seen in ex vivo cultures of hemocytes,25 depletion of apoptosomal factors rescued cells from ethanol-induced cytotoxicity but, surprisingly, alcohol-metabolizing enzymes did not. Using this platform, we conducted a genome-wide RNA interference (RNAi) screen to identify effectors that support ethanol-induced cell death. One of these effectors, Drat (Death Resistor Adh domain containing Target), is a novel protein that contains an ADH domain, but lacks essential residues in its catalytic site. Depletion of Drat protected distinct cell lines from alcohol challenge and, when tested in vivo, adult neurons depleted of Drat were similarly protected from ethanol toxicity. Animals mutated for Drat showed improved survival after alcohol challenge and also exhibited enhanced propensities toward alcohol-induced sedation. Taken together, these studies identify apoptogenic effectors triggered by alcohol and highlight phenotypic consequences resulting from interventions that prevent cell deaths caused by this stressor.
Results
Depletion of the apoptosome protects cells from ethanol-induced apoptosis
Using an ex vivo culture system, we previously showed that ‘loss-of-function’ genetics could be applied to examine alcohol-induced cell killing in Drosophila. Specifically, we found that hemocytes aspirated from animals lacking either Dark or Dronc were fully resistant to ethanol exposures that were otherwise toxic to wild-type hemocytes.25 As shown in Figure 1, we generalized these findings to the Drosophila S2 cell line. When exposed to ethanol, these cells exhibited an apoptogenic response similar to that of larval hemocytes,25 but if the apical caspase Dronc (Figures 1d–h) was silenced, death was prevented as assessed by two distinct readouts for viability, Sytox green (measures membrane permeability properties,26 Figure 1f) and CellTiter-Glo (measures ATP levels,27 Figure 1g). At lower ethanol concentrations (1% or less), Dronc silencing completely rescued cells from ethanol-induced apoptosis. At higher ethanol concentrations (2%), only partial rescue was observed. In both instances, control dsRNAs (double-stranded RNAs) targeting irrelevant genes had no effect. Silencing of Dark, the adapter component necessary for apoptosome formation,28 similarly rescued cells from ethanol-induced killing (Figure 4).

Dronc is required for alcohol-induced cell death. Micrographs of S2 cells untreated (a) and ethanol-treated (b) illustrates that alcohol challenge provokes extensive apoptosis, shown here 48 h postexposure to 2% ethanol (arrows in b and c indicate characteristic apoptotic cells and corpses). When cells are pretreated with irrelevant dsRNA (against GFP), ethanol challenge provokes widespread apoptosis (c). However, if parallel cultures are similarly pretreated with Dronc dsRNA, apoptosis is prevented (d). These data are corroborated and quantified in panels (f) and (g), where two different cell viability measures are used. (f) Control and Dronc dsRNA-treated cells were treated with indicated ethanol concentrations for 24 h and cell viability is measured using the Sytox assay (Sytox Green, Molecular Probes/Life Technologies, Carlsbad, CA, USA). (f and g) Cell viability is measured using the CellTiter-Glo assay (see Materials and Methods). In both panels, cell survival (%) was normalized to control cells not treated with ethanol (mean±S.D., n=3). Dronc RNAi consistently reversed ethanol-induced killing, assayed here under two different treatment protocols. In these experiments, 20 000 cells were plated in wells that contained 0.5 μg of Dronc or GFP dsRNA, incubated for 2 (f) or 3 days (g) before 48 h of ethanol treatment. Under both protocols, silencing Dronc effectively prevents cell death. (e) Semiquantitative RT-PCR is shown validating effective silencing of Dronc RNA. Detection of rp49 here shows specificity of silencing. (h) Retention of proliferative capacity in ‘rescued’ cells. Here, S2 cells were incubated with Dronc or GFP dsRNA for 3 days before re-plating at equal density followed by a 24-h challenge as indicated (in the presence of dsRNAs). Cells were then recovered and cultured in fresh media without ethanol for an additional 3 days. Relative growth in this post-treatment phase was determined by comparing ethanol challenged samples with untreated controls (average of two trials). Together, these studies establish that silencing expression of Dronc prevents apoptosis and preserves cell viability in the context of alcohol challenge
To further confirm these findings, we assessed the proliferative capacity of RNAi-treated cells after ethanol exposure in ‘wash-out’ experiments. Figure 1h shows that Dronc RNAi-treated cells retained significantly greater proliferative capacities after ethanol was withdrawn as compared with negative controls. These findings establish that, if certain apoptogenic functions are removed, cells can survive subacute levels of ethanol challenge. In addition, these results showed that the function of apoptosome genes, Dronc and Dark, is required for ethanol-induced cell death in Drosophila cells.
To determine whether these findings might generalize to other cell types, we tested other Drosophila cell lines, and found that ethanol also triggered apoptosome-dependent death of Kc 167 cells. In particular, Kc 167 cells pretreated with ecdysone exhibited a robust Dronc-dependent response with a broad dynamic range. Differentiated Kc 167 cells also ceased dividing after treatment with this hormone,29 enabling direct assessment of cytotoxicity without confounding effects relating to cell growth. Therefore, Kc 167 cells were used in many subsequent assays.
Given that silencing of apoptosomal genes protects cells from ethanol-induced cell death, we systematically tested all Drosophila caspases, for similar protective activity. As the two effector caspases, Dcp1 and Drice, can function redundantly we also tested the combined effects of dsRNAs targeting both enzymes. When tested singly or in combination, as shown in Figure 2 and reported elsewhere,25, 30 Dcp1 and Drice were clearly ‘rate-limiting’ for cell death in the context of UV challenge (Figure 2b). In contrast, neither of these effector caspases were rate-limiting in the context of ethanol challenge even when both were simultaneously targeted (Figure 2a). Furthermore, a systematic study of all Drosophila caspases found that none, except Dronc, were rate-limiting under these conditions (Figure 2a). Together, these data suggest that killing by UV and killing by ethanol are distinct at the effector caspase level.

Requisite caspase profiles provoked by ethanol are distinct. Kc 167 cells treated with indicated caspase dsRNAs, control dsRNAs and vehicle (water) were challenged with 1% ethanol (a) or UV (b). Cell survival was measured with CellTiter-Glo and normalized to untreated cells (mean±S.D., n=3). For drice and strica, two distinct dsRNA amplicons (kd1 and kd2) were used to target these genes
Ethanol-induced cell death is unaffected by suppression of Adh and Adhr genes
Ethanol-metabolizing enzymes, including Adh and Aldh, are frequently implicated in models of alcohol-induced tissue injury.31, 32 To directly test if Drosophila Adh and Aldh genes are important for alcohol-induced apoptosis, we silenced Adh, Adhr (Adh-related) and Aldh genes with appropriate dsRNAs before ethanol exposure and assayed for protection from (or enhancement of) cell killing. As shown in Figure 3a, neither Adh nor Adhr dsRNAs, alone or in combination, modified ethanol-induced killing. Alcohol-induced death was similarly unaffected by silencing of Aldh (Figure 3b). As Adh and Aldh enzymes initiate alcohol catabolism, these results indicate that ethanol, rather than a derived metabolite, likely constitutes the apoptogenic stimulus in this system.

Ethanol-induced cell death is unaffected by suppression of alcohol-metabolizing enzymes. (a) Kc 167 cells treated with dsRNAs targeting luciferase (an irrelevant control), Dronc, Adh and/or Adhr and vehicle (water) were challenged with 1% ethanol. (a, inset) Semiquantitative RT-PCR detects relative transcript levels of Adh and Adhr in dsRNA-treated cells as indicated. (b) Kc 167 cells treated with control dsRNAs targeting ampicilin-resistant gene (AmpR), Dronc, Aldh (two amplicons, kd1 and kd2) and vehicle (water) were challenged with 1% ethanol. (b, inset) Semiquantitative RT-PCR detects relative transcript levels of Aldh in dsRNA-treated cells as indicated. In both panels, cell survival was measured with CellTiter-Glo and normalized to untreated cells (mean±S.D., n=3)
Genome-wide RNAi screen identified effectors of ethanol-induced cell death
To identify effectors required for ethanol-induced cell death, we optimized parameters for a genome scale RNAi screen. Toward this goal, we assayed S2 cells and Kc 167 cells, under various conditions. As they produced a broad dynamic range with superior consistencies in high throughput formats, differentiated Kc 167 cells were used to conduct a genome scale screen. Figure 4 illustrates how this platform, together with a library of dsRNAs targeting 13 017 annotated genes (Drosophila genome build 3), was used to identify genes that suppress or enhance ethanol-induced cell death. Each dsRNA was tested in triplicate and each assay plate included four wells of Dronc dsRNAs as a positive control (see Materials and Methods for details). Using both plate and position mean-centering analyses, we identified 30 candidate genes with protective activity at a z-score ≥2.8 threshold. This collection includes the expected benchmark genes Dronc and Dark as well as four other targets identified in a previous screen for apoptotic modifiers (hrb27c, myb, ef2b, atpα).33 Of the remaining 24 candidates, all were retested using a different dsRNA amplicon to exclude off-target effects33 (see Materials and Methods for details). Six of these were retested with a z-score of 3.0 or above. In addition, 40 protective candidates with z-scores between 2.1 and 2.8 were retested, 3 of which gave z-scores above 3.0. A total of 10 candidates with enhancing activity (at z-score ≤−2.5) were also retested and two of these (CG9811 and CG9375) showed reproducible activity at a z-score of –3.0. Thus, as summarized in Table 1, a total of nine dsRNAs with suppressor activity and two dsRNAs with enhancing were confirmed after retesting. One enhancer (CG9811), was previously known to be required for cell viability34 and was not pursued further.

A genome-wide screen captures effectors of ethanol-induced cell death. (a) Schematic for the screen protocol. See Materials and Methods for details and statistical analyses. Note that each dsRNA was scored in triplicate ( × 3). (b) Average position mean-centered z-scores for the genome-wide screen are plotted in rank format. Arrowheads indicate ‘landmark’ apoptotic genes and the alcohol-metabolizing enzymes Adh/Adhr and Aldh
Drat is an effector of alcohol-induced cell death in vitro and in vivo
To distinguish whether candidates recovered here encode specific effectors of ethanol-induced cell death, or whether they function more generally in apoptosis, each was tested in the context of UV-induced cell killing.30 As seen in Table 1, three genes were protective against both ethanol challenge and UV exposure when silenced (ate1, CG32579, eIF4G). Six targets blocked ethanol-induced death but were inactive in UV-treated cells. When silenced, CG9375, a validated enhancer, was also specifically involved in ethanol-induced cell death.
Among the genes found to be specifically required for ethanol-induced apoptosis, one target, CG1600, encoded a member of the zinc-dependent ADH-like family, which was reported responsive to alcohol in a previous study.35 Within its ADH homology domain, CG1600 has an N-terminal catalytic domain with distant homology to GroES and a C-terminal NAD(P) binding-Rossmann fold domain. Importantly, although CG1600 contains a putative NAD(P)-binding site, none of the Cys or His residues needed for alcohol binding or catalytic activity are present (NCBI Conserved Domain Database). Therefore, the product of this gene does not encode a canonical Adh enzyme. As shown in Table 1, silencing this gene protected Kc 167 cells from ethanol-induced death, but did not protect against UV treatment. Therefore, we designated this gene Drat. To examine if depletion of Drat might impact other cell types, we performed similar experiments using S2 cells. As seen in Figure 5, comparable protection against ethanol-induced killing is seen both S2 and Kc 167 cell lines.

Drat is required for alcohol-induced cell death in both Kc cells and S2 cells. Kc 167 cells (a) or S2 cells (b) in 96-well microplates treated with 1.0 μg dsRNAs targeting luciferase or ampicilin, Dronc, Drat or vehicle (no dsRNAs) were challenged with 2% ethanol for 3 days, and cell survival was measured with CellTiter-Glo. Two distinct non-overlapping dsRNAs targeting Drat (kd1 and kd2) were tested here. Data was normalized to cells that were not challenged with ethanol (mean±S.D., n=3). (a, inset) Semiquantitative RT-PCR detects relative transcript levels of Drat and rp49 in cells treated with dsRNA in parallel as in the experiment
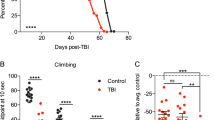
To examine Drat function in vivo, we tested Drat− animals for defective responses to alcohol exposure. For this purpose, we characterized a transposon strain P{PTT-GB}CG1600CB03410 (CB03410) corresponding to an insertion in the first intron of the Drat locus (see Materials and Methods). At least three transcripts are produced from this locus (Drat-RA, -RB and -RC). As seen in Figures 6a and b, Drat-RC is the major isoform, whereas Drat-RA and Drat-RB are expressed at relatively low levels. In CB03410 homozygous mutants, levels of the minor Drat-RB isoform were not overtly affected but expression levels of Drat-RC and Drat-RA were severely impaired (Figures 6a and b). We examined these hypomorphic flies using assays that measure whole animal survival and apoptotic cell death after exposure to alcohol vapor (see Materials and Methods). Compared with wild-type controls sharing the identical genetic background (see Materials and Methods), male and female CB03410 mutants showed substantially increased survival after exposure (Figure 6c).

Drat is an effector of alcohol-induced cell death in vivo. (a) RT-PCR of wild-type and P{PTT-GB}CG1600CB03410-mutant adults. Primers that are specific for each transcript of Drat (CG1600) were used to detect levels of Drat-RA, -RB and -RC. Note that in P{PTT-GB}CG1600CB03410, RA and RC fragments are larger than their wild-type counterparts by virtue of a transposon insertion. As the RB-specific primer spans the wild-type splicing junction, it failed to detect a GFP fusion transcript in P{PTT-GB}CG1600CB03410. The RC-GFP fragment (RC*) was sequenced to confirm the GFP insertion. (b) RT-PCR of homozygous P{PTT-GB}CG1600CB03410 and wild-type adults using primer pairs diagnostic for the RB and RC transcript isoforms in wild-type and CB03410 flies. RC* denotes Drat-RC-GFP fragment. (c) Three-day-old P{PTT-GB}CG1600CB03410 and wild-type adult flies were treated with ethanol vapor for 1 h, and survival was assessed after 24 h (see Materials and Methods). Data shown are the averages from five experiments, using a total of more than 700 adults. Error bars indicate standard deviation. (d) Three-day-old P{PTT-GB}CG1600CB03410 and wild-type adult flies were treated with ethanol vapor for 30 min and blackening of the third antennal segment was quantified the next day as in French and Heberlien.36 Data shown are averages from two independent trials. (e and f) Images of heads from control and ethanol-challenged Or83b-Gal4/UAS-RFP flies. The third segment of antenna (arrows) and maxillary palps (arrowheads) were labeled with RFP and killing of olfactory neurons by ethanol exposure was assessed by following loss of RFP in the third antennal segment as in French and Heberlien.36 The dotted boxes highlight RFP signal in the antenna and maxillary palps. Note that (f) is overexposed to appreciate RFP loss, resulting in nonspecific signal from the eyes indicated by *. (g) Note that ethanol-induced death of olfactory neurons cells is partially rescued by silencing of Drat. Flies expressing RFP and indicated dsRNAs under the control of Or83b-GAL4 were exposed to ethanol for 30 min, and the percentage of unaffected RFP-positive antenna (arrows) was scored the next day (mean±S.D., n=4). Inset: RT-PCR from flies expressing various dsRNA constructs driven by da-GAL4 confirms effective silencing of Drat by the relevant transgenes. In these experiments, two distinct Drat dsRNA transgenes (drat-1 and drat-2) and dsRNAs against two irrelevant genes CG8184 and CG7926 were used. (h) P{PTT-GB}CG1600CB03410 and P{PTT-GB}CG1600CB03410/+ flies were challenged with ethanol vapor, and median sedation time (ST50) was measured (mean±S.D., n=3; see Materials and Methods)
Recently, French and Heberlien36 showed that blackening of the third antennal segment is a correlate for apoptotic death of olfactory receptor neurons after exposure to ethanol vapor. To investigate whether Drat functions to support ethanol-induced apoptosis in vivo, we scored this phenotype in adults the day following ethanol exposure, and as shown in Figure 6d, substantially fewer CB03410 mutants showed this alcohol-induced phenotype. Apoptotic loss of olfactory neurons can also be directly quantified by using the Or83b-Gal4 ‘driver’ to express a red fluorescent protein (RFP) reporter in these cells36 (Figures 6e and f). Therefore, we combined this reporter system together with dsRNA-silencing transgenes to directly assess whether Drat contributes to the loss of these neurons after ethanol exposure. For this purpose, we validated two distinct transgenes that silence Drat (Figure 6g, inset) and the effects of these dsRNA transgenes (drat-1 and drat-2) were compared to others from this same collection that target irrelevant genes (CG8184 and CG7926). As seen in Figure 6g, depletion of Drat in olfactory neurons afforded considerable protection to these cells after challenge by ethanol vapors. Taken together, these results establish that Drat function is required for ethanol-induced cell death in vitro and in vivo. Furthermore, the results also establish a positive correlation between apoptotic resistance to this stressor at the cellular level and resistance to this stressor at the whole animal level.
To investigate whether elimination of Drat might also impact ethanol-associated behaviors, we tested these animals in a loss-of-righting assay,37 which is frequently used to assess propensities toward sedation. As seen in Figure 6h, CB03410 homozygotes become sedated more rapidly than their heterozygous counterparts, indicating a role for Drat in this behavior.
Discussion
The precise mechanisms that link alcohol consumption to pathologic cell death are largely unknown. Although apoptosis is well established as a critical pathologic feature contributing to ethanol-induced tissue injury, molecular effectors that couple alcohol exposure to this and other regulated forms of cell death are not known. In fact, most of our knowledge regarding pathways implicated in alcohol-induced apoptosis come from reports that correlate alcohol exposure to changes in expression or activity.19
We postulated signaling events that specify apoptotic responses in the alcohol-challenged cell and tested this prediction using the Drosophila system. As seen previously with mutant hemocytes lacking the apical caspase Dronc,25 silencing of either Dronc or its adapter Dark, similarly blocked ethanol-induced cell death in cultured cells. These results provide a compelling example whereby single-gene ablation can preclude alcohol-induced cell killing. Our findings also establish an obligate role for the apoptosome in this model system and demonstrate that when core apoptogenic functions are removed, cells can in fact survive chronic ethanol challenge. Notably, there was a sharp concentration threshold for rescue by RNAi (Figure 1) enabling an operational distinction between apoptogenic signaling (at lower doses) and nonspecific cytotoxicity (at higher doses). Furthermore, as genetically rescued cells were able to proliferate once ethanol was removed (Figure 1h), these genetic interventions also suggest that cellular damage encountered during subacute alcohol exposure can be reversible.
Our findings also suggest a likely scenario whereby ethanol instigates unknown signals that somehow engage the apoptosome. Surprisingly, however, silencing effector caspases, including drice and dcp1, alone or in combination, failed to influence ethanol-induced death despite the fact that both were clearly rate-limiting for death after UV exposure (Figure 2). Consistent with this result, no caspase other than Dronc emerged as a suppressor in our genome-wide dsRNA-silencing screen (Figure 4). Hence, although the apoptosome was clearly essential no single effector caspase was rate-limiting for alcohol-induced apoptosis. Therefore, downstream of the apoptosome, multiple effector caspases could be equally engaged or, alternatively, perhaps caspase-independent pathway(s) are involved.
What actually instigates apoptosis in the alcohol-challenged cell? It is well established that Adh and Aldh influence responses to alcohol stress through intermediates of oxidative metabolism.7, 38 Conversion of ethanol to acetaldehyde by these enzymes is commonly proposed as a mediator of ethanol-associated cytotoxicity (reviewed in Dey and Cederbaum4 and Hoek and Pastorino5) and, therefore, if acetaldehydes are responsible for instigating apoptosis, depletion of Adh should protect from cell death and depletion of Aldh should enhance cell killing. Here, we directly tested both the predictions and, in both cases, the outcome was unexpected. Ethanol-induced cell killing was unaffected by depletion of Adh and Adhr alone or in combination (Figure 3) and, equally surprising, Aldh depletion failed to enhance the apoptogenic effects of ethanol. Moreover, even under relaxed stringencies, none of these canonical alcohol-metabolizing enzymes produced significant z-scores as suppressors or enhancers of cell death in our genome scale analysis (Figure 4). Taken together, these results suggest that ethanol itself, rather than an Adh-derived metabolite, is the agent that instigates apoptosis under the conditions used here.
To identify additional effectors, we completed a genome-wide RNAi screen for modifiers of ethanol-induced cell death. Among the ‘hits’ found here, we note that two definitive benchmarks, Dark and Dronc, as well as four genes identified in a previous screen25 for apoptotic effectors triggered by an IAP antagonist were recovered. The fact that these landmark genes were captured assures a high-level quality control and confirms that the screen performed as expected. In addition, after layered rounds of retesting a collection of 10 high-confidence targets was recovered, including 9 suppressors and 1 enhancer. These genes may encode general apoptogenic effectors or specific effectors that couple ethanol stress to this response. To empirically distinguish between these possibilities, each target was additionally tested in a model UV-induced death and all but three conferred activity specific to alcohol challenge (Table 1). The remaining seven genes with activities selective to ethanol stress were queried using computational programs to detect enriched biological or molecular processes. Although no specific effector pathways were identified at high significance, one effector drew attention as a computed gene with a highly relevant domain (CG1600). This gene, designated Drat, encodes products containing an N-terminal ADH GroES-like domain (ADH_N domain), and a C-terminal domain corresponds to an NAD(P)-binding-Rossmann fold. Residues critical for NAD(P) binding are present in Drat but Cys or His residues thought to be essential in the active site are clearly absent. Therefore, despite containing domains that enable NADP binding, Drat may not be an enzyme and could plausibly function as a sensor of alcohol levels in exposed cells. Given its potential for alcohol binding, future studies will test this possibility and examine whether it encodes non-canonical activity. Interestingly, Drat was moderately induced after ethanol treatment in flies35 but was not sufficient to enhance ethanol-induced killing when over-expressed (P Chen, X Tu and J Abrams, unpublished observations). Whatever its mechanism of action, depletion of Drat protected cultured cell lines and adult neurons in vivo from alcohol-induced apoptosis. Therefore, the action of Drat extends well beyond the original cell type used in our screening platform and, as seen here, can be exploited for in vivo protection of neurons in animals that have been challenged.
Finally, in the whole animal assays, adults hypomorphic for Drat were more easily sedated and less likely to die after exposure to ethanol fumes. Additional studies are needed to understand how resistance to apoptosis at the cellular level might translate to these whole animal phenotypes. Nevertheless, our data suggest that behavioral propensities toward sedation could be an adaptive response that aids survival in the context of this class of stressors.
Materials and Methods
Cell culture, ecdysone differentiation and ethanol treatment
S2 and Kc 167 cells were cultured in Schneider’s media with 10% fetal bovine serum, 25 U/ml penicillin, 25 μg/ml streptomycin at 25°C (all cell culture reagents from Invitrogen/Life Technologies (Carlsbad, CA, USA) except FBS (Atlas Biologicals, Fort Collins, CO, USA)). Unless otherwise indicated, media in all procedures refer to the above. Drosophila cells treated with two dsRNAs were exposed to 15 μg/ml of each indicated dsRNA, for a total dsRNA concentration of 30 μg/ml. For ecdysone differentiation, Kc 167 cells were plated in CCM3 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10 μM ecdysone (Axxora, San Diego, CA, USA). After 3 days, the media was removed, and replaced with CCM3 containing 1% ethanol. Two days later, cell survival was assessed with CellTiter-Glo (Promega, Madison, WI, USA) according to manufacturer’s instructions. To obtain % survival, raw data from treated samples were normalized to the averaged mean of untreated control cells.
Primary and secondary RNAi screen
The primary screen was optimized for Kc 167 cells and conducted using a ‘direct soaking’ protocol, together with the Silencer Drosophila RNAi library (Ambion/Life Technologies (Carlsbad, CA, USA)) targeting 13 071 genes from Drosophila genome build 3. Sequences for all dsRNAs in the library are available in Chew et al.33 The RNAi library or synthesized dsRNAs were plated in 96-well microplates (Corning, Lowell, MA, USA) using Beckman FX liquid handlers. Every assay plate included four control wells containing Dronc dsRNA. Each well contained 0.5 μg dsRNA in 20 μl of SF900 media. A total of 18 000 cells in 30 μl CCM3 media were seeded in each well and incubated for 1 h before adding 50 μl CCM3 media containing 20 μM ecdysone. Three days later, the media was replaced with 80 μl of 1% ethanol CCM3 media. Cell viability was assayed 2 days later using CellTiter-Glo (Promega) in a plate reader (Envision multimode, Perkin Elmer, Waltham, MA, USA). The plate mean centered z-score for each well is its luminescence value minus the plate average, divided by the plate standard deviation (n=92 sample wells). The Z-factor for each plate was calculated as described.39 Low quality (Z<0) plates were excluded from subsequent secondary retests and analyses. To correct for systematic bias/edge effects,40 the position mean centered z-score for each well is calculated by its plate mean centered z-score minus the position average, divided by the position standard deviation (n=143 plates per triplicate). Average position mean centered z-score for each amplicon is calculated from the individual position mean centered z-scores of the triplicates. Genes with z-score >2.8 from either plate mean or position mean centered normalizations were considered together as primary candidates. For secondary screens and subsequent RNAi experiments, dsRNAs synthesis and treatment were as described33 (Supplementary Table 1). For each candidate target we synthesized two distinct dsRNAs (Supplementary Table 1). Assays in undifferentiated Kc 167 and S2 cells were performed following the same procedure as described above, except that ecdysone was not added, 1.0 μg dsRNA was present in each 96-well microplate and 2% ethanol was used. For RNAi of caspases, the transcript levels of dronc, drice, dcp-1 and dredd were quality controlled by RT-PCR to assure effective silencing.
Drosophila strains and assays
Or83b-Gal4, Da-Gal4 and UAS-stinger Red flies are from Bloomington Stock Center (Bloomington, IN, USA). Flies carrying dsRNA transgenes targeting various genes were obtained from the Vienna Drosophila RNAi Center (Vienna, Austria). The P{PTT-GB}CG1600CB03410 strain was kindly provided by Dr. Buszczak. We verified this strain by sequencing an RT-PCR fragment corresponding to the Drat-RC transcript. In P{PTT-GB}CG1600CB03410 flies, we confirmed that green fluorescent protein (GFP) is fused in frame with Drat-RC, replacing the first 16 residues of this transcript. The P{PTT-GB}CG1600CB03410 insertion was crossed into the wberlin background for 10 generations before phenotypic analyses. This same background corresponds to wild type shown in Figures 6a–d. All assays (except loss-of-righting assay) used 20–25 flies that were 3 days posteclosion at the beginning of the experiment. For survival assays, flies were exposed to ethanol vapor for 60 min and numbers of dead and live flies were counted 24 h later. For antennal assays, flies were exposed to ethanol vapor for 30 min and the numbers of normal and blackened antenna, or the number of RFP-positive and -negative antenna, were counted 24 h later. For loss-of-righting assay, flies were exposed to ethanol, and ST-50 was determined as described.37
Abbreviations
- FAS:
-
fetal alcohol syndrome
- ROS:
-
reactive oxygen species
- ADH:
-
alcohol dehydrogenase
- ALDH:
-
acetaldehyde dehydrogenase
- Adhr:
-
ADH-related
- GALT:
-
gut-associated lymphoid tissue
- Drat:
-
Death Resistor ADH domain containing Target
- ORNs:
-
olfactory receptor neurons
- GFP:
-
green fluorescent protein
- RFP:
-
red fluorescent protein
- dsRNA:
-
double-stranded RNA
- RNAi:
-
RNA interference
References
US Department of Health and Human Services Tenth Special Report to the US Congress on Alcohol and Health. DHSS, PHS-NIAAA: US Government Printing Office, 2000.
Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000; 287: 1056–1060.
Baker RC, Kramer RE . Cytotoxicity of short-chain alcohols. Annu Rev Pharmacol Toxicol 1999; 39: 127–150.
Dey A, Cederbaum AI . Alcohol and oxidative liver injury. Hepatology 2006 43 (Suppl 1: S63–S74.
Hoek JB, Pastorino JG . Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 2002; 27: 63–68.
Shukla SD, Sun GY, Gibson Wood W, Savolainen MJ, Alling C, Hoek JB . Ethanol and lipid metabolic signaling. Alcohol Clin Exp Res 2001; 25 (Suppl ISBRA): 33S–39S.
Hoek JB, Cahill A, Pastorino JG . Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 2002 122: 2049–2063.
Asai K, Buurman WA, Reutelingsperger CP, Schutte B, Kaminishi M . Low concentrations of ethanol induce apoptosis in human intestinal cells. Scand J Gastroenterol 2003; 38: 1154–1161.
Slomiany BL, Piotrowski J, Slomiany A . Chronic alcohol ingestion enhances tumor necrosis factor-alpha expression and salivary gland apoptosis. Alcohol Clin Exp Res 1997; 21: 1530–1533.
Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ et al. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ 2003; 10: 1148–1155.
Souza HS, Elia CC, Braulio VB, Cortes MQ, Furtado VC, Garrofe HC et al. Effects of ethanol on gut-associated lymphoid tissues in a model of bacterial translocation: a possible role of apoptosis. Alcohol 2003; 30: 183–191.
Guerri C . Mechanisms involved in central nervous system dysfunctions induced by prenatal ethanol exposure. Neurotox Res 2002; 4: 327–335.
Canbay A, Gieseler RK, Gores GJ, Gerken G . The relationship between apoptosis and non-alcoholic fatty liver disease: an evolutionary cornerstone turned pathogenic. Z Gastroenterol 2005; 43: 211–217.
Canbay A, Friedman S, Gores GJ . Apoptosis: the nexus of liver injury and fibrosis. Hepatology 2004; 39: 273–278.
Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ . Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol 2001; 34: 248–253.
Yoon JH, Gores GJ . Death receptor-mediated apoptosis and the liver. J Hepatol 2002; 37: 400–410.
Purohit V, Brenner DA . Mechanisms of alcohol-induced hepatic fibrosis: a summary of the Ron Thurman Symposium. Hepatology 2006; 43: 872–878.
Aroor AR, Shukla SD . MAP kinase signaling in diverse effects of ethanol. Life Sci 2004; 74: 2339–2364.
Pani G, Fusco S, Colavitti R, Borrello S, Maggiano N, Cravero AA et al. Abrogation of hepatocyte apoptosis and early appearance of liver dysplasia in ethanol-fed p53-deficient mice. Biochem Biophys Res Commun 2004; 325: 97–100.
Hoek JB, Pastorino JG . Cellular signaling mechanisms in alcohol-induced liver damage. Semin Liver Dis 2004; 24: 257–272.
Guarnieri DJ, Heberlein U . Drosophila melanogaster, a genetic model system for alcohol research. Int Rev Neurobiol 2003; 54: 199–228.
Wolf FW, Heberlein U . Invertebrate models of drug abuse. J Neurobiol 2003; 54: 161–178.
Rodan AR, Kiger JA, Heberlein U . Functional dissection of neuroanatomical loci regulating ethanol sensitivity in Drosophila. J Neurosci 2002; 22: 9490–9501.
Hay BA, Guo M . Caspase-dependent cell death in Drosophila. Annu Rev Cell Dev Biol 2006; 22: 623–650.
Chew SK, Akdemir F, Chen P, Lu WJ, Mills K, Daish T et al. The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev Cell 2004; 7: 897–907.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1: 112–119.
Petty RD, Sutherland LA, Hunter EM, Cree IA . Comparison of MTT and ATP-based assays for the measurement of viable cell number. J Biolumin Chemilumin 1995; 10: 29–34.
Rodriguez A, Oliver H, Zou H, Chen P, Wang XD, Abrams JM . Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat Cell Biol 1999; 1: 272–279.
Gauhar Z, Sun LV, Hua S, Mason CE, Fuchs F, Li TR et al. Genomic mapping of binding regions for the Ecdysone receptor protein complex. J Biolumin Chemilumin 2009; 19: 1006–1013.
Zimmermann KC, Ricci JE, Droin NM, Green DR . The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol 2002; 156: 1077–1087.
Guo R, Ren J . Alcohol dehydrogenase accentuates ethanol-induced myocardial dysfunction and mitochondrial damage in mice: role of mitochondrial death pathway. PLoS One 2010; 5: e8757.
Li SY, Gomelsky M, Duan J, Zhang Z, Gomelsky L, Zhang X et al. Overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene prevents acetaldehyde-induced cell injury in human umbilical vein endothelial cells: role of ERK and p38 mitogen-activated protein kinase. J Biol Chem 2004; 279: 11244–11252.
Chew SK, Chen P, Link N, Galindo KA, Pogue K, Abrams JM . Genome-wide silencing in Drosophila captures conserved apoptotic effectors. Nature 2009; 460: 123–127.
Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B et al. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 2004; 303: 832–835.
Morozova TV, Anholt RR, Mackay TF . Transcriptional response to alcohol exposure in Drosophila melanogaster. Genome Biol 2006; 7: R95.
French RL, Heberlein U . Glycogen synthase kinase-3/Shaggy mediates ethanol-induced excitotoxic cell death of Drosophila olfactory neurons. Proc Natl Acad Sci USA 2009; 106: 20924–20929.
Rothenfluh A, Threlkeld RJ, Bainton RJ, Tsai LT, Lasek AW, Heberlein U . Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell 2006; 127: 199–211.
Marchitti SA, Brocker C, Stagos D, Vasiliou V . Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol 2008; 4: 697–720.
Zhang JH, Chung TD, Oldenburg KR . A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999; 4: 67–73.
Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R . Statistical practice in high-throughput screening data analysis. Nat Biotechnol 2006; 24: 167–175.
Acknowledgements
We would like to thank UTSW HTS Lab and Dr. Lum for invaluable advice and support for dsRNA screens. This work was supported by grants to JMA from the National Institute on Alcohol Abuse and Alcoholism (R01AA017328), the National Institute of General Medical Sciences (R01GM072124) and the Welch Foundation (Grant No. I-1727) and by the grant R01AA019526 to AR.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Chen, P., Tu, X., Akdemir, F. et al. Effectors of alcohol-induced cell killing in Drosophila. Cell Death Differ 19, 1655–1663 (2012). https://doi.org/10.1038/cdd.2012.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.47
Keywords
This article is cited by
-
Overexpression of a novel F-box protein PsFFL1 from tree peony (Paeonia suffruticosa) confers drought tolerance in tobacco
Plant Growth Regulation (2023)
-
Altering social cue perception impacts honey bee aggression with minimal impacts on aggression-related brain gene expression
Scientific Reports (2019)
-
Molecular mechanisms of hepatic apoptosis
Cell Death & Disease (2014)
