
Abstract
Eukaryotic cells are equipped with an efficient quality control system to selectively eliminate misfolded and damaged proteins, and organelles. Abnormal polypeptides that escape from proteasome-dependent degradation and aggregate in the cytosol can be transported via microtubules to inclusion bodies called ‘aggresomes’, where misfolded proteins are confined and degraded by autophagy. Here, we show that Type 2 transglutaminase (TG2) knockout mice display impaired autophagy and accumulate ubiquitinated protein aggregates upon starvation. Furthermore, p62-dependent peroxisome degradation is also impaired in the absence of TG2. We also demonstrate that, under cellular stressful conditions, TG2 physically interacts with p62 and they are localized in cytosolic protein aggregates, which are then recruited into autophagosomes, where TG2 is degraded. Interestingly, the enzyme's crosslinking activity is activated during autophagy and its inhibition leads to the accumulation of ubiquitinated proteins. Taken together, these data indicate that the TG2 transamidating activity has an important role in the assembly of protein aggregates, as well as in the clearance of damaged organelles by macroautophagy.
Similar content being viewed by others
Main
Misfolded proteins are a constant byproduct of eukaryotic cell metabolism resulting from genetic mutations, inappropriate posttranslational protein modifications, and environmental stress. It has been proposed that up to 30% of newly synthesized proteins are not properly folded.1 These non-functional misfolded proteins are prone to form aggregates that can interfere with normal cellular functions inducing cellular stress, which can ultimately lead to cell death. Thus, the efficient management of misfolded proteins and their aggregates is essential for cellular viability, and thus cells have evolved several interconnected pathways to limit their accumulation.2 This system of protein quality control acts at multiple levels, trying to re-establish the proper tertiary structure of unfolded or misfolded proteins via molecular chaperones, and eventually promoting the degradation of those aberrant proteins that fail to refold.2 In fact, the misfolded proteins are recognized and poly-ubiquitinated by specific ubiquitin E3 ligases and this posttranslational modification targets them to proteasome degradation, which constitutes a critical part of the cytoprotective protein quality control machinery.3, 4 Defects in the ubiquitin–proteasome machinery lead to the accumulation of misfolded proteins and aggregates under the specific form of ‘aggresomes’, a pathological condition commonly found in many degenerative diseases.2 Recent findings suggest that the formation of aggresomes is an attempt to prevent the intracellular toxicity derived from protein aggregates before they are actively removed by autophagy.5 In fact, once aggregated, misfolded proteins cannot be degraded efficiently by proteasome as they cannot pass through the narrow structures of the proteasome.6
Autophagy was initially described as a cellular response to nutrient deprivation by which cells are able to ‘self-digest’ part of their own cytoplasm to recycle essential macromolecules for survival.7 In the most characterized form of autophagy, macroautophagy, starvation induces a double-membrane structure that sequesters cytosolic contents (including mitochondria, peroxisomes, and ribosomes) into large autophagic vesicles to form the autophagosomes. Autophagosomes subsequently fuse to lysosomes, where their contents are degraded by hydrolytic enzymes.8 This feature makes autophagy an ideal way to remove protein aggregates that cannot be processed by proteasomes. The molecular mechanisms regulating the specific recognition and elimination of the aggresomes by autophagy have just recently begun to be uncovered.
This process requires autophagy cargo proteins, such as p62/SQSTM1 and NBR1, which bind ubiquitinated proteins via UBA domains, and interact with LC3 via their LIR domain.9 Therefore, at least two modifications, ubiquitination and oligomer formation, have been proposed to serve as cis-acting signals to promote selective autophagy.
Type 2 transglutaminase (TG2) is a multifunctional protein that is capable of carrying a large number of biochemical functions. In the presence of high calcium levels, TG2 catalyzes a vast array of protein posttranslational modifications, including protein–protein crosslinking, incorporation of primary amines into proteins, as well as glutamine deamination. In addition, TG2 might also act as a G-protein, coupling various receptors to phospholipase C (PLCδ1).10 This activity is inhibited by Ca2+, which then acts as a switch between the two main biochemical functions of the enzyme.10 Under physiological conditions, the low cytosolic Ca2+ concentration does not allow the enzyme to act as a transamidating agent.11 However, catalytically inactive TG2 can perform other functions; for example, TG2 can associate with integrins to activate signaling pathways contributing to increase cell survival.10 If the stress or cellular damage is too severe, the Ca2+-dependent activation of TG2 crosslinking activity has an important role in the induction of apoptosis.12
Several studies show that TG2 is involved in the pathogenic mechanisms responsible for a number of human degenerative diseases characterized by the accumulation of protein aggregates in affected brains, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and in liver diseases (Mallory bodies).13, 14 We have recently shown that TG2 is involved in autophagosome maturation.15, 16 Starting from these premises we decided to analyze the role of TG2 in autophagy and shed light on the possible involvement of the enzyme in the degenerative disorders involving the formation of aggresomes and their clearance by autophagy.
Results
Loss of TG2 leads to a defective autophagic response in the liver and heart upon starvation
In order to get an insight into TG2's role in the autophagic process, we decided to investigate the effect of its ablation in vivo by producing a transgenic mouse line lacking TG2 and expressing GFP-LC3, which is widely used as an in vivo marker for autophagic vesicles. The newly developed transgenic mouse line shows no abnormality and reproduces normally.
Liver
Considering the importance of autophagy in the liver upon starvation, we decided to start our analysis by examining this organ.17 In both TG2+/+ GFP-LC3- or TG2−/− GFP-LC3-fed mice, GFP-LC3 signals were found to diffuse in the cytoplasm with few punctate dots (Figures 1a and c). Following 48 h of starvation (Starv), the number of GFP-LC3 dots increased in both mouse strains, indicating the expected induction of autophagy. However, in the TG2−/− mice, the accumulation of the dots was significantly higher than in TG2+/+ mice (Figures 1b and d), suggesting that, in the absence of TG2, more autophagosomes are formed and/or less autophagosomes are degraded by fusing with lysosomes. The quantification of GFP-LC3-positive dots confirmed the increase of these structures after starvation (Figure 1e), with a significant difference in their number in livers of TG2−/− mice compared with the TG2+/+ mice. These results are in agreement with the impairment of autophagosome maturation observed in vitro in cells lacking TG2.15

Effect of starvation on liver and heart autophagy in TG2+/+ GFP-LC3 or TG2−/− GFP-LC3 mice. (a and b) Liver samples from TG2+/+ GFP-LC3 transgenic mice before (a) and after 48 h of starvation (b) were analyzed by fluorescence microscopy. (c and d) Liver samples from TG2−/− GFP-LC3 transgenic mice before (c) and after 48 h of starvation (d) were analyzed by fluorescence microscopy. Scale bar, 8 μm. (e) Quantitative analysis of the formation of GFP-LC3 dots following autophagy-induction. The number of dots was counted in five independent fields of three independent mice. Results are expressed as the mean±S.D. Statistical analysis was performed by analysis of variance (*P<0.05). (f and g) Heart samples from TG2+/+ GFP-LC3 transgenic mice before (f) and after 48 h of starvation (g) were analyzed by fluorescence microscopy. (h and i) Heart samples from TG2−/− GFP-LC3 transgenic mice before (h) and after 48 h of starvation (i) were analyzed by fluorescence microscopy. Scale bar, 3 μm. (j) Quantitative analysis of the formation of GFP-LC3 dots following autophagy-induction. The number of dots was counted in five independent fields of three independent mice. Results are expressed as the mean±S.D. Statistical analysis was performed by analysis of variance (*P<0.05, **P<0.001, ***P<0.0001)
Heart
It is well known that heart muscle shows major changes during starvation; in fact, the number and size of autophagosomes increased drastically after 48 h of fasting.17 Therefore, we decided to compare autophagy levels also in cardiac muscles of TG2+/+ GFP-LC3 with those of TG2−/− GFP-LC3 mice following 48 h of starvation. As illustrated in Figures (1f–i), the increase in GFP-LC3-positive structures was more accentuated in TG2−/− mice compared with the TG2+/+ mice. The quantification of the GFP-LC3 dots evidenced a drastic difference between the levels of autophagy induced in TG2+/+ mice compared with TG2−/− mice (Figure 1j) similarly to those described above for the liver.
TG2 is degraded during autophagy
On the basis of the above-reported in vivo findings, we decided to further address the role of TG2 in autophagy. To this aim, we analyzed the TG2 protein level in mouse liver after 24–48 h of starvation. Interestingly, we observed a progressive significant decrease (up to 70% at 48 h) in the total protein levels compared with normally fed animals (Figure 2a). To verify whether this phenomenon could be due to the enzyme degradation into autophagolysosomes, we treated the starved animals with chloroquine (CQ), which is known to block the acidification of lysosomes, thus preventing the last stages of autophagy. The results reported in Figure 2b indicate that TG2 is rescued from degradation when the lysosomal activity is partially reduced by CQ. In order to verify whether the degradation of TG2 was a general autophagy-dependent event, we induced autophagy in 2fTGH cells, a human cell line expressing high TG2 levels, by several agents, that is, starvation, 2-deoxy-D-glucose (2-DG) and rapamycin (Rap) (Figure 2c). In the presence of all tested autophagy stimuli we observed a significant decrease of TG2 protein levels, which was comparable to that of p62, an autophagy-cargo protein degraded inside autophagolysosomes (unpublished observations). Prompted by this result, we investigated whether TG2 was detectable inside autophagosomes, by analyzing its co-localization with LC3 upon autophagy induction. The immunofluorescence analysis carried out in 2fTGH-GFP-LC3 cells reported in Figures 3a and b shows that the two proteins co-localize in punctate vesicular structures upon starvation. In keeping with this finding, ultrastructural analysis showed the presence of TG2 immuno-gold positive particles inside double-membrane autophagosomes (Figures 3c and d). To confirm the autophagosomal nature of these structures, we co-immunostained the same sections for p62, which showed the same localization (Figure 3d).

TG2 is degraded by autophagy. (a, upper panel) Immunoblot analysis of TG2 in liver tissues from TG2+/+ and TG2−/− mice, before and after 24 and 48 h of starvation. Actin was used as loading control. (a, lower panel) Densitometric analysis of blots (quantification of TG2 bands normalized to actin levels). (b, upper panel) Immunoblot analysis of TG2 in liver tissues from TG2+/+ mice, before and after 24 and 48 h of starvation in the presence or not of CQ. For the autophagy inhibition the CQ was administered in the drinking water during the whole starvation time. Actin was used as loading control for immunoblot. (b, lower panel) Densitometric analysis of blots (quantification of TG2 bands normalized to actin levels). TG2 expression is reduced following autophagy induction and is rescued from degradation when the lysosomal activity is blocked by CQ. (c, upper panel) Western blot analysis of TG2 in 2fTGH cells subjected to different treatments to induce autophagy (see Materials and Methods). Actin was used as loading control for immunoblot. (c, lower panel) Densitometric analysis of blots (quantification of TG2 bands normalized to actin levels)

TG2 colocalizes with LC3 in the autophagosomes. (a and b) Immunofluorescence microscopy analysis of 2fTGH-GFP-LC3 cells undergoing autophagy. Cells expressing GFP-LC3 protein were cultured in complete medium (a) or subjected to starvation (b) in EBSS for 16 h, stained with an anti-TG2 antibody and analyzed by microscopy. Green dots represent autophagosomes, red dots TG2 protein, and yellow dots sites of overlap of GFP-LC3 with TG2. Scale bar, 6 μm. (c and d) Immuno-gold analysis in human 2fTGH cells double-labeled against TG2 (15-nm gold particles) and p62 (5-nm gold particles). The colloidal gold particles, indicating the presence of TG2 (arrows) and p62 (arrowheads), were detected in typical autophagosome-like structures. Original magnification: × 85 000
TG2 is present in protein aggregates containing p62
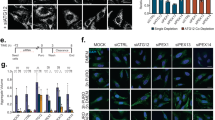
Several reports have suggested that TG2 has a role in the formation of ubiquitinated protein aggregates found in diseases affecting both the brain and the liver.18.These TG2-related aggregates contain poly-ubiquitinated proteins and therefore are likely to be bound by p62, via its UBA domain.9 In order to verify whether TG2 can physically interact with the cargo proteins mediating the delivery of ubiquitinated proteins to autophagosomes, we analyzed by immunoprecipitation the interaction of the enzyme with p62, NBR1 and the valosin-containing protein (VCP), which are essential elements able to convey ubiquitinated substrates into the autophagosomes.19 To this aim, we used 2fTGH cells, HEK293 cells in which the TG2 expression is undetectable, and HEK293TG2, in which the wild-type enzyme was stably ectopically expressed (Supplementary Figure S1A). The results indicate that TG2 is part of a protein complex containing p62 (Figures 4a and b) and NBR1 (Supplementary Figure S1B), while the interaction with VCP (Figures 4a and b) and LC3 (unpublished observations) was not detected. Interestingly, in both cell lines the interaction between TG2 and p62 was largely increased upon the inhibition of the proteasome by MG132, which is known to determine the accumulation of ubiquitinated protein aggregates (Figure 7f).20 In order to clarify whether TG2 cooperates with the cargo proteins in the clearance of ubiquitinated proteins, we analyzed TG2 transamidating activity in 2fTGH cells upon starvation. To this aim, we used the 5-(biotinamido)pentylamine incorporation method to verify the enzyme's transamidating activity in vivo under the employed experimental conditions. Intracellular TG2 is known to be largely inactive under normal physiological settings, but it is activated under stressful conditions, which lead to the accumulation of free calcium in the cytoplasm.11 Interestingly, we detected an increased labeling of TG2 substrate proteins already upon autophagy induction, but the amine incorporation was dramatically increased when autophagy induction was associated with the proteasome inhibition (Figure 4c).

TG2 interacts with autophagy-related cargo proteins. (a) Western blot analysis of TG2, p62 and VCP proteins in human 2fTGH cells subjected to immunoprecipitation for TG2. After 4 h of treatment as indicated, cells were lysed and proteins were immunoprecipitated using anti-TG2 antibody (see Materials and Methods). Immuno- and co-immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted using the indicated antibodies. WC, whole cell lysate, was used as protein control. (b) Western blot analysis of TG2, p62 and VCP proteins in HEK293TG2 cells stably transfected with TG2 protein, subjected to immunoprecipitation for TG2. After 2 h of treatment as indicated, cells were lysed and proteins were immunoprecipitated using anti-TG2 antibody. Immuno- and co-immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted using the indicated antibodies. WC, whole cell lysate, was used as protein control. (c) Representative blot of the TG-catalyzed incorporation of 5-(biotinamido)pentylamine into proteins in 2fTGH cell line. Cells were labelled with 5-(biotinamido)pentylamine and treated as indicated for 4 h. After separation by SDS-PAGE, biotinylated proteins were revealed with HRP-conjugated streptavidin. p53 and p27 proteins were used as a control for monitoring proteasome inhibition, and actin was used as loading control
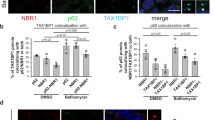
The blockade of proteasomal degradation is known to determine a stressful cellular condition leading to the formation of ‘aggresomes’ containing ubiquitinated proteins.20 To determine whether, under these conditions, TG2 and p62 co-localize on the aggresomes, we carried out the analysis of their ultrastructural localization in starved cells treated with MG132. The data reported in Figure 5 indicate that the enzyme is specifically localized in protein aggregates of various sizes. Interestingly, the co-localization with p62 is observed only in protein aggregates, which display a large size, thus suggesting that TG2 could be recruited on small protein aggregates and participates to form larger complexes where p62 is later recruited. Furthermore, the immunofluorescence co-localization analysis of p62 and TG2, in cells treated with MG132, confirmed the presence of both proteins in the aggresomes (Supplementary Figure S2).

TG2 is present in cytosolic protein aggregates. (a and b) HEK293TG2 cells were cultured under starved condition in presence of MG132 for 2 h. Cells were double-labelled against TG2 (15-nm gold particles), and against p62 (5 nm gold particles). Immuno-gold analysis shows that small cytosolic membrane-free electron-dense structures (arrowheads) are positively stained for TG2 while larger aggregates display simultaneous positivity for TG2 and p62 (arrows). Original magnification: × 85 000
TG2 is involved in pexophagy
Considering that p62-dependent autophagy of ubiquitinated proteins has been shown to have a key role also in the clearance of damaged and hyper-accumulated organelles, such as mitochondria and peroxisomes, we decided to verify whether the above-described defect in TG2-mediated autophagy could be reflected in an abnormal homeostatic control of these organelles.19 In fact, ubiquitination of peroxisomal proteins in mammalian cells is sufficient to cause their p62-mediated turnover by targeting these organelles to autophagosomes and lysosomes.21
We took advantage of a well-established pharmacological protocol of peroxisomal induction and subsequent removal in liver rodents.22, 23 Peroxisomes are known to increase in their number and size, in response to a class of chemically unrelated substances, called peroxisome proliferators; when their administration is discontinued, the liver peroxisomal population rapidly decreases and readily returns to the pre-proliferation status due to autophagy-mediated clearance.21, 24 We chose a model of inducing peroxisomal proliferation by dietary administration of ciprofibrate. Excess liver peroxisomes are normally degraded within a week, but this rapid removal is impaired in autophagy-deficient liver.25
Morphological and molecular data indicated effectiveness of the treatment on both TG2−/− and TG2+/+ mouse liver (Figure 6). Remarkable peroxisomal proliferation was demonstrated by increased number of catalase-positive organelles, as detected by transmission electron microscopy, immediately after ciprofibrate treatment (T0; Figures 6a and b). Consistently, the levels of the 70-kDa peroxisomal membrane protein (PMP70) were significantly higher in treated animals than in controls (Figure 6e). At later stages (T7), the size of peroxisomal population returned to normal values in the TG2+/+ liver, as assessed by PMP70 western blot analysis (Figure 6e), while in the hepatocytes of TG2−/− mice, numerous peroxisomes were still present, as detected by ultrastructural (Figures 6c and d) and western blot analyses (Figure 6e). Though often found inside autophagic vacuoles, these organelles displayed normal morphological features, including homogeneous catalase cytochemical activity, and electron-dense cores (Figure 6d). These hallmarks, indicating intactness of the organelles in the knockout liver, were paralleled by intense PMP70 immunoreactivity, significantly higher than in TG2+/+ samples (Figure 6e).

Impaired in vivo pexophagy after ciprofibrate-induced peroxisomal proliferation in the liver of TG2−/− mice. Ultrastructural catalase cytochemistry of TG2+/+ (a) and TG2−/− (b–d) mouse liver, after ciprofibrate treatment. Mice were given ciprofibrate-containing food for 10 days, then killed immediately (T0) or 7 days (T7) after suspension of treatment. Liver specimens were prepared as described in Materials and Methods and ultrathin sections were observed in a Zeiss EM 900 electron microscope. (a and b) Remarkable peroxisomal induction is observed in both TG2+/+ and TG2−/− hepatocytes, at T0. However, while autophagosomes with partially degraded material are readily detected in the TG2+/+ samples (arrows), the knockout shows virtually no autophagic vacuoles. (c and d) Several autophagosomes are recognized in TG2−/− samples at T7. These mostly contain indigested peroxisomes, with homogeneous catalase positivity (arrowheads). The higher magnification micrograph (d) shows the details of an autophagic vacuole (arrowhead), surrounding an intact peroxisome, in which the double membrane is clearly visible, catalase is cytochemically active and a typical peroxisomal core (white arrowhead) is present. N, nucleus; m, mitochondrion; p, peroxisome. Original magnification: (a and b) × 12 000; (c × 4400;) (d × 20 000.) (e and f) Densitometric analysis of western blotting data, obtained on liver extracts from TG2+/+ and TG2−/− mice before and after in vivo ciprofibrate administration, using the peroxisomal marker PMP70 (e) and the autophagic marker LC3 normalized to GADPH (f). LC3 levels are expressed as the ratio between LC3 II and LC3 I. Statistical analysis was performed by analysis of variance (*P<0.05)
Consistent with morphological data, immunoblot results using the autophagic marker LC3 II (Figure 6f) showed dramatic induction of the autophagic program in TG2+/+ liver, soon after suspension of ciprofibrate in vivo administration (T0). Later on, LC3 levels returned rapidly to control values (T7). By contrast, LC3 levels in TG2−/− liver were unchanged at T0, and raised in the first week after treatment, indicating, together with the increased level of PMP70, a much slower clearance of peroxisomes due to an impaired autophagy flux.
Interestingly, autophagy serves to degrade intracellular lipid stores in murine liver through a process that has been termed ‘macrolipophagy’.26 Histological examination of paraffin sections stained either by hematoxylin and eosin (H&E) or by Sudan Black, as well as ultrastructural analysis, demonstrates lipid accumulation in TG2−/− mouse liver after ciprofibrate treatment (Supplementary Figure S3), further confirming the impairment of autophagy in the presence of a defect in TG2 transamidating activity.
Impaired clearance of ubiquitinated protein aggregates in the absence of TG2 transamidating activity
Based on the above-described results, we decided to verify whether the defect of autophagy induced by the lack of TG2 in vivo was characterized by the accumulation of ubiquitinated protein aggregates.
To this end, we analyzed the liver of TG2 knockout and wild-type mice upon starvation. Under fed condition some ubiquitin-positive cells were found in TG2+/+ mice (Figures 7a and e). However, the amount of ubiquitinated protein aggregates was much more pronounced in TG2-deficient mouse (Figures 7b and e). Interestingly, following 48 h of starvation, the number of cells with ubiquitinated protein aggregates decreased in TG2+/+ mouse, as a result of their clearance by autophagy (Figures 7c and e). On the contrary, in the liver of TG2−/− mouse, an increased number of cells with ubiquitinated protein aggregates was observed (Figures 7d and e), thus suggesting that, in the absence of TG2, elimination of protein aggregates is affected. In order to demonstrate that the accumulation of ubiquitinated proteins observed in knockout animals was indeed dependent on the lack of TG2, we analyzed the profile of ubiquitinated proteins in HEK293 cells lacking the enzyme and in the same cells overexpressing TG2 protein (HEK293TG2). The data reported in Figure 7f clearly show that in the absence of the enzyme there was a marked accumulation of high-molecular-weight ubiquitinated proteins, particularly evident when the proteasome inhibition was carried out by MG132.

Effect of TG2 on ubiquitinated aggregates accumulation. (a–d) Immunofluorescence detection of ubiquitin in the liver of TG2+/+ and TG2−/− mice before (a and b) and after starvation (c and d). Scale bar, 8 μm. (e) Quantitative analysis of the formation of ubiquitin-positive aggregates in basal condition and after autophagy induction. The number of cells containing aggregates was counted in five independent fields of three independent mice. Results are expressed as the mean±S.D. Statistical analysis was performed by analysis of variance (*P<0.05, **P<0.001). (f) Immunoblotting analysis of ubiquitinated proteins in HEK293 and HEK293TG2 cell lines. Cells were incubated with or without MG132 for 2 h, and ubiquitin expression levels were assayed by immunoblotting with anti-ubiquitin antibody. Actin was used as loading control
To characterize whether this effect is dependent on the TG2 crosslinking activity, we analyzed the importance of the enzyme's transamidating activity in the clearance of ubiquitinated protein aggregates. To this aim, we studied the direct interaction of ubiquitinated proteins with TG2 using the HEK293 cell line stably transfected with the wild-type enzyme and with its mutant lacking the transamidating activity (HEK293C277S) (Supplementary Figure S1A). Interestingly, the active TG2 and its transamidating mutant both interact with high-molecular-weight ubiquitinated proteins (Figure 8a), thus indicating that the transamidating activity is dispensable for the binding of ubiquitinated proteins. On the contrary, the transamidating activity is essential for their clearance, as shown by the accumulation of high-molecular-weight ubiquitinated proteins detected in cells expressing the C277S mutant (Figure 8b). Furthermore, the data reported in Figure 8a suggest that the interaction between TG2 and the ubiquitinated proteins is not dependent on p62, as protein complexes co-immunoprecipitating with the TG2 mutant contain significant less amount of this cargo protein.

Effect of TG2 transamidating activity on the clearance of ubiquitinated protein aggregates. (a) Western blot analysis of p62 and ubiquitinated proteins in HEK293, HEK293TG2 and HEK293C277S cell lines subjected to immunoprecipitation for TG2. Cells treated with MG132 for 2 h were lysed and proteins were immunoprecipitated using anti-TG2 antibody. Immuno- and co-immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted using the indicated antibodies. Arrows indicate ubiquitinated proteins present in TG2 interacting protein complexes. (b) Immunoblotting analysis of ubiquitinated proteins in HEK293, HEK293TG2 and HEK293C277S cell lines. Cells were incubated with MG132 for 2 h, and ubiquitin expression levels were assayed by immunoblotting with anti-ubiquitin antibody. Actin was used as loading control
Discussion
The data reported in this study indicate that TG2 is an essential component for the proper maturation of autophagosomes under basal and particularly under stressful cellular conditions such as proteasome impairment or organelle hyper-proliferation. Indeed, we have previously shown that the ablation of TG2 results in an evident accumulation of LC3 II on pre-autophagic vesicles, pointing to a marked induction of autophagy.15 By contrast, the formation of the acidic vesicular organelles was very limited, as detected by the lack of acidification of the autophagolysosomes observed in TG2−/− MEFs, suggesting an impairment in the autophagosome maturation process.15 In this paper, we demonstrate that the ablation of TG2 leads to the accumulation of ubiquitinated organelles and protein aggregates, which are not cleared by autophagy, thus indicating a role for the enzyme in the selection of the autophagy substrates. In fact, in the absence of TG2 and particularly under stressful conditions, we observed both in vivo and in vitro a marked accumulation of ubiquitinated proteins. We have also shown that the TG2-dependent posttranslational modification of its substrate proteins is increased by autophagy induction, and further potentiated upon proteasome inhibition. In connection with this, it is accepted that during the early stage of autophagy there is an accumulation of free calcium ions in the cytoplasm that may activate the TG2's transamidating activity.27
Interestingly, TG2 is present in protein complexes containing two cargo proteins, p62 and NBR1, which are known to mediate the clearance of ubiquitinated proteins by autophagy. These cargo proteins contain UBA domains at their C-terminal regions, allowing them to interact with the aggregated protein complexes and organelles that have to be degraded. Subsequently, by a LIR domain these cargo proteins interact with the ATG8-family members inside the nascent pre-autophagic vesicles.9, 19 It is interesting to note that, although TG2 has two potentially canonical LIR domains, we were unable to detect by immunoprecipitation an interaction between TG2 and LC3 (unpublished observations). By contrast, our findings indicate that TG2 is localized in small protein aggregates where p62 is not present, while this cargo protein is later recruited on large aggregates where a high amount of TG2 is detected. In keeping with this finding, we detected an increased co-localization of the two proteins upon inhibition of the proteasome, a condition known to favor the formation of ‘aggresomes’.20 Furthermore, in cells lacking TG2, we detected the accumulation of ubiquitinated proteins suggesting that the enzyme is involved in the posttranslational modification of high-molecular-weight aggregates, which are then conveyed by cargo proteins to the autophagic machinery for degradation. Notably, only the reintroduction of the transamidating active TG2 in these cells favors the clearance of ubiquitinated proteins, highlighting that TG2's crosslinking activity has an important role in the formation/stabilization of substrates cleared by the autophagy pathway. These findings are in agreement with our previous results showing that the transamidating activity of the enzyme is required for the proper completion of the autophagic flux15, 16 and the inhibition of the transamidating activity of TG2, by means of the specific inhibitor R283, results in a marked decrease of the number of acidic vesicular organelles.15 In this regard, it is important to mention that TG2 has been involved in the pathogenesis of several degenerative diseases characterized by the accumulation of mutated/misfolded proteins such as Lewy bodies in PD, neurofibrillary tangles in AD, Huntingtin aggregates in HD, as well as the Mallory bodies in the liver.13, 14 Our previous studies, carried out in a mouse model for HD, suggested a possible involvement of TG2 in the neurodegenerative processes associated with autophagy impairment observed in this disease. In fact, the analysis of brains of the TG2−/−/HD transgenic mice showed that the HD onset is associated with a large reduction in non-apoptotic cell death and with an increased number of nuclear protein inclusions, suggesting an impairment in their clearance by autophagy.28
Taken together, these data indicate an important role for TG2-mediated posttranslational modifications in the maturation of autophagosomes. In support of this assumption is also the long-lasting use of a well-known competitive inhibitor of transglutaminase, the auto-fluorescent compound mono-dansyl-cadaverine (MDC), for in vivo labelling of autophagic vacuoles. MDC accumulates as a selective marker for autophagic vacuoles in vivo, and is not present in the early and late endosome.29 In keeping with this, we have shown here that TG2 is present inside the autophagosomes, where the enzyme is selectively degraded similarly to its binding partner p62.
It is well known that autophagy has a crucial role in the turnover of cellular organelles; in particular it has been proposed that autophagy selectively degrades dysfunctional or supernumerary mitochondria and peroxisomes. Here, we show that pexophagy following ciprofibrate treatment is impaired in mice lacking TG2. Also, we have recently shown that the absence of TG2 is associated with the impairment of mitochondria that, in light of the present observations, could be due to the lack of clearance of the damaged ones.13
Finally, it is relevant to note that a large number of characterized TG2 candidate protein substrates interact with the ATG8 family members,30 thus providing further evidence that the TG2-catalyzed posttranslational modifications may have a role in their clearance through the autophagosomes in collaboration with p62 and NBR1. The TG2 transamidating activity, acting at the level of glutamine and lysine residues of proteins, might be envisaged as a further regulatory step in the selection of protein substrates to be degraded into autophagolysosomes. In line with this assumption, it is important to mention that so far no clear consensus sequence has been described for the TG2's substrate proteins. Several authors have proposed that glutamine and lysine residues well exposed on the protein surface can act as possible targets for the enzyme, a condition likely to occur in misfolded proteins.31 On the basis of the data presented in this study we would like to propose that TG2 can be involved in the early steps of the recognition of the ubiquitinated proteins and organelles to be degraded. TG2 could favour by its transamidating activity the formation of intracellular structures, which are recognized by p62 and other cargo proteins leading to their recruitment in the pre-autophagic vesicles before degradation by autophagolysosomes (Figure 9).

Tentative scheme of TG2 action during autophagy. The accumulation of misfolded ubiquitinated proteins, sensed by the cell as a stress, leads to the activation of autophagy at ER level determining the release of free calcium ions in the cytoplasm (1). This event leads to the activation of TG2 transamidating activity associated with its 3D structural change from the inactive ‘closed conformation’ to the active ‘opened conformation’ (2). The activated enzyme may catalyze the misfolded protein crosslinking, leading to the formation of small aggregates (3) and subsequently large aggregates (4). The formation of these ubiquitinated inclusions is recognized via the UBA domains by p62 or other cargo proteins. These complexes finally interact with LC3II through the LIR domains present on the cargo proteins, leading to their uploading inside the pre-autophagic vesicles
Materials and Methods
Antibodies
Anti-tubulin, anti-actin and anti-PMP70 were from Sigma-Aldrich (Saint Louis, MO, USA), anti-LC3 was from Novus Biologicals (Littleton, CO, USA), anti-TG2 TG100 or CUB7402 were from Lab Vision NeoMarkers (Fremont, CA, USA), anti-mono and poly-ubiquitinated conjugates were from Enzo (Plymouth Meeting, PA, USA), anti-p62/SQSTM1 was from MBL (Woburn, MA, USA), anti-VCP, anti-p27 (C-19), anti-p53 (FL-393) and anti-GADPH (FL-335) were from Santa Cruz (Santa Cruz, CA, USA), and anti-NBR1 was from Cell Signaling (Danvers, MA, USA). HRP-conjugated secondary antibodies were from Bio-Rad Laboratories (Hercules, CA, USA). Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies were from Invitrogen (Carlsbad, CA, USA).
Generation of TG2−/− GFP-LC3 mice
In order to obtain TG2−/− mice expressing GFP-LC3 (TG2−/− GFP-LC3), C57Bl/6 TG2−/− mice were crossed with C57Bl/6 GFP-LC3 transgenic mice. TG2−/− genotype was assessed by PCR using 5′-TCCTGACCTGAGTCCTCGTC-3′, 5′-TACTCCAGCTTCTCGTTCTG-3′, and 5′-ACGAGACTAGTGAGACGTGC-3′ primers, as previously described.32 To assess the presence of GFP-LC3 transgene, PCR was carried out with:
Primer 1 (5′-ATAACTTGCTGGCCTTTCCACT-3′)
Primer 2 (5′-CGGGCCATTTACCGTAAGTTAT-3′)
Primer 3 (5′-GCAGCTCATTGCTGTTCCTCAA-3′)
(intron 3 of the LC3 genome as an internal control) following the conditions 94°C for 5 min, then 40 cycles at 94°C for 30 s+55°C for 30 s+72°C for 1 min, and then 72°C for 5 min.
Cell culture, transfection and drug treatments
HEK293 (human embryonic kidney) and 2fTGH were cultured in Dulbecco's modified Eagle's medium (Invitrogen Life Technologies Inc.) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 mg/ml streptomycin and 100 units/ml penicillin in a 5% CO2 incubator. For generation of the TG2 stably expressing cells (HEK293TG2 and HEK293C277S), HEK293 cells were transfected with pLPCX-TG2 or pLPCX-C277S16 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions and then selected for puromycin (Sigma-Aldrich) resistance using the antibiotic added to the culture medium (2 μg/ml). For the generation of 2fTGH-GFP-LC3 cells, 2fTGH cells were transiently transfected with pLPCX-GFP-LC3 vector using Lipofectamine 2000.
For amino-acid starvation (Starv), cells were washed two times in Earle's balanced salt solution (EBSS; Sigma-Aldrich) and incubated in EBSS for the indicated periods in the presence or not of 5 μM MG132 (Z-Leu-Leu-Leu-al; Sigma-Aldrich). For autophagy induction cells were incubated in full medium in the presence of 1 μM Rap (Sigma-Aldrich) or 10 mM 2-DG (Sigma-Aldrich).
Animals and treatment
Animals were fed with 0.025% ciprofibrate (Sigma Chemical Co., St. Louis, MO, USA). Mice were given 40 g ciprofibrate-containing food for 10 days, then killed immediately (T0) or 7 days (T7) after suspension of treatment.
For the autophagy induction mice were fed ad libitum or subjected to 24 or 48 h of starvation. For the autophagy inhibition the CQ was administered in the drinking water. Considering that the water uptake of mice is between 4 and 7 ml per day, we calculated a concentration of CQ salt of 0.375 mg/ml dissolved in normal tap water to deliver a daily oral dose of 1.5–2.6 mg.33
Immunoblotting
Tissues were lysed in HEMG buffer (25 mM Hepes pH 8.0, 100 mM NaCl, 25 mM MgCl2, 0.5% Triton X-100, 0.1 mM EDTA, and 10% glycerol) supplemented with protease and phosphatase inhibitor cocktail (1 mM sodium fluoride, 1 mM sodium orthovanadate and 1 mM sodium molybdate; Sigma-Aldrich).
Cells were rinsed in ice-cold phosphate-buffered saline (PBS) and collected in lysis buffer, containing 20 mM Tris-HCl pH 7.4, 150 mM NaCl and 1% Triton X-100 with protease inhibitor cocktail (Roche, Nutley, NJ, USA).
Protein concentrations were determined by the Bradford assay, using bovine serum albumin as a standard.
Aliquots of total protein extracts from cells and tissues after different treatments were resolved on SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. Blots were blocked in 5% non-fat dry milk in T-PBS (PBS+0.05% Tween 20) for 1 h at room temperature and then incubated overnight with the above-described antibodies. The membranes were incubated with HRP-conjugated secondary antibody for 1 h at room temperature and the signal was detected by Immun-Star WesternC Kit (Bio-Rad Laboratories).
Statistical analysis
Densitometry measurements were used for statistical analysis using Graph Pad. Statistical significance was determined using the Student's t-test. A P-value ⩽0.05 was considered to be significant.
Fluorescence microscopy
Tissues were washed by cardiac perfusion using PBS, dissected and fixed in 4% paraformaldehyde (PFA). Tissues were then cryoprotected with increasing concentrations of sucrose (15 and 30%, overnight) in phosphate buffer. Next, tissues were transferred to an embedding mold filled with Tissue–Teck OCT compound. The mold was rapidly submerged into isopentane cooled. The material was then frozen and stored at −80°C. The samples were sectioned at 10–20 μm thickness with cryostat (ref ex: CM3050 S, Leica, Deerfield, IL, USA) and stored at −80°C. For GFP observation, nuclei were stained with DAPI (20 min, 1 μg/ml) and then cryosections were examined under a Leica TCS SP5 confocal microscope equipped with an × 40 (NA 1.25) or × 63 (NA 1.4) oil-immersion objective (Deerfield, IL, USA). We used ‘Leica confocal’ software (Deerfield, IL, USA) for analysis. For immunohistochemical analysis, sections were permeabilized with 0.4% Triton X-100 in PBS for 1 h and then blocked in 3% NGS in HSPBS containing 0.2% Triton X-100 for 1 h. Sections were incubated with the above-indicated primary antibody.
Subconfluent cells (2fTGH and 2fTGH-GFP-LC3) grown on coverslips were washed with PBS and fixed in 4% PFA for 10 min at room temperature. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min, blocked with 3% BSA in PBS for 20 min, and incubated with primary antibodies for 1 h.
After washing, cells and tissue sections were incubated with Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies. The coverslips were mounted on microscope slides, sealed with an antifade solution and examined with an image workstation DeltaVision (Applied Precision, Issaquah, WA, USA) Olympus 1X70 microscope.
Quantitative analysis of GFP-LC3 dots and ubiquitin-positive aggregates
The number of GFP-LC3 dots in the liver and heart was counted in five independent fields from three independent mice for each condition. Each point value represents the mean±S.D. Statistical analysis was performed by analysis of variance (*P<0.05).
The number of ubiquitin-positive inclusions was counted in five independent fields from three independent mice for each condition. Each point value represents the mean±S.D. Statistical analysis was performed by analysis of variance (*P<0.05).
Histological examination
Tissues were dissected, fixed in 4% PFA, paraffin embedded, and sectioned. Sections were stained by Meyer's hematoxylin and eosin.
Sudan Black (Sigma-Aldrich) staining was performed on cryostat liver sections, after dehydration in 70% ethanol for 30 min. Freshly prepared saturated Sudan Black solution was prepared in 70% ethanol. Sections were incubated with Sudan Black solution for 10 min, then treated with 70% ethanol for 1 min, and finally mounted using crystal mount (Biomeda, Burlingame, CA, USA).
Catalase ultrastructural cytochemistry
Liver specimens were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, for 45 min at 4°C. After extensive rinse in the same buffer, samples were incubated for 1 h at 37°C in the dark in 0.05 M 2-amino-2-methyl-1,3-propanediol, pH 9.4, containing 7.5% sucrose, 5 mM NaCN, 0.2% 3,3′-diaminobenzidine-tetra HCl (DAB) and 0.05% H2O2. For controls, the incubation medium was supplemented with 0.1 M 3-amino-1,2,4-triazole.
Specimens were then rinsed in 0.1 M cacodylate buffer, pH 7.4, postfixed in 1% OsO4 in the same buffer for 1 h, at 4°C, dehydrated, and embedded in epoxy resin.
All the specimens were cut on a Leica UltraCut S ultramicrotome (Leica Microsystems, Wetzlar, Germany). Semithin sections were stained with toluidine blue and photographed, and ultrathin sections were briefly contrasted with aqueous 2% uranyl acetate for 20 min. Grids were observed and photographed in a Zeiss EM 900 electron microscope (Zeiss, Oberkochen, Germany).
Immunoprecipitation
Cell lysates were prepared in HEMG buffer (25 mM Hepes pH 8.0, 100 mM NaCl, 25 mM MgCl2, 0.5% Triton X-100, 0.1 mM EDTA, and 10% glycerol) and protease inhibitor cocktail (Roche). The lysates were subjected to immunoprecipitation using specific antibodies in a combination with Protein G PLUS–Agarose (Santa Cruz). Immunoblot analysis was performed with indicated antibodies.
In situ TG assay
In situ TG activity was quantified by determining the incorporation of 5-(biotinamido)pentylamine into protein substrates. In brief, cells were pre-incubated for 20 min with 2 mM Ez-link pentylamine-Biotin (Thermo Scientific, Rockford, IL, USA) before each treatment. Cells were then harvested, lysed as above described, and proteins were resolved by SDS polyacrylamide gel. The blots were probed with streptavidin HRP-conjugated (Amersham Biosciences, Little Chalfont, UK) for 2 h at room temperature and then developed following the standard ECL protocol.
Immunoelectron microscopy
For immunolabelling, cells were fixed in 4% PFA, dehydrated in ethanol, and embedded in LR-White resin. Ultrathin sections were mounted on Formvar-coated copper grids, then washed three times with PBS and three times with PBS containing 1% bovine serum albumin and 0.15% glycine, followed by 30 min blocking with 5% normal goat serum. Samples were incubated overnight with the primary antibodies, mouse anti-TG2 1 : 25, and rabbit anti-p62 1.3 : 100. After washing in PBS, samples were incubated for 45 min with goat anti-mouse IgG conjugated to 15 nm gold particles and goat anti-rabbit IgG conjugated to 5 nm gold particles (BioCell, Cardiff, UK). Sections were stained with 2% uranyl acetate and observed under a Zeiss EM900 transmission electron microscope. Images were captured digitally with a Mega View II digital camera (Olympus SIS, Munster, Germany). The specificity of the immunoreaction was assessed in all cases by omitting the primary antibodies from the labelling protocol and incubating the sections only in the protein gold-conjugated secondary antibodies.
Abbreviations
- TG2:
-
Type 2 transglutaminase
- Starv:
-
starvation
- CQ:
-
chloroquine
- 2- DG:
-
2-deoxy-D-glucose
- Rap:
-
rapamycin
- MG132:
-
Z-Leu-Leu-Leu-al
- VCP:
-
valosin-containing protein
- PMP70:
-
peroxisomal membrane protein
- HD:
-
Huntington's disease
- MEFs:
-
mouse embryonic fibroblast
References
Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR . Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000; 404: 770–774.
Yao TP . The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer 2010; 1: 779–786.
Cyr DM, Hohfeld J, Patterson C . Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci 2002; 27: 368–375.
Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C . Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res 2010; 106: 463–478.
Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 2003; 12: 749–757.
Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B . Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem 2003; 278: 11753–11759.
Klionsky DJ, Emr SD . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721.
Klionsky DJ . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8: 931–937.
Lamark T, Kirkin V, Dikic I, Johansen T . NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8: 1986–1990.
Fesus L, Piacentini M . Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci 2002; 27: 534–539.
Lai TS, Bielawska A, Peoples KA, Hannun YA, Greenberg CS . Sphingosylphosphocholine reduces the calcium ion requirement for activating tissue transglutaminase. J Biol Chem 1997; 272: 16295–16300.
Fesus L, Thomazy V, Autuori F, Ceru MP, Tarcsa E, Piacentini M . Apoptotic hepatocytes become insoluble in detergents and chaotropic agents as a result of transglutaminase action. FEBS Lett 1989; 245: 150–154.
Malorni W, Farrace MG, Rodolfo C, Piacentini M . Type 2 transglutaminase in neurodegenerative diseases: the mitochondrial connection. Curr Pharm Des 2008; 14: 278–288.
Strnad P, Zatloukal K, Stumptner C, Kulaksiz H, Denk H . Mallory-Denk-bodies: lessons from keratincontaining hepatic inclusion bodies. Biochim Biophys Acta 2008; 1782: 764–774.
D’Eletto M, Farrace MG, Falasca L, Reali V, Oliverio S, Melino G et al. Transglutaminase 2 is involved in autophagosome maturation. Autophagy 2009; 5: 1145–1154.
Rossin F, D’Eletto M, Macdonald D, Farrace MG, Piacentini M . TG2 transamidating activity acts as a reostat controlling the interplay between apoptosis and autophagy. Amino Acids 2011. doi:10.1007/s00726-011-0899-x (in press).
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y . In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101–1111.
Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina DL, Settembre C et al. Cystic fibrosis: a disorder with defective autophagy. Autophagy 2011; 7: 104–106.
Weidberg H, Shvets E, Elazar Z . Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011; 80: 125–156.
Guthrie CR, Kraemer BC . Proteasome inhibition drives HDAC6-dependent recruitment of tau to aggresomes. J Mol Neurosci 2011; 45: 32–41.
Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J . Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 2008; 105: 20567–20574.
Fahimi HD, Reinicke A, Sujatta M, Yokota S, Ozel M, Hartig F et al. The short- and long-term effects of bezafibrate in the rat. Ann NY Acad Sci 1982; 386: 111–135.
Islinger M, Luers GH, Li KW, Loos M, Volkl A . Rat liver peroxisomes after fibrate treatment: a survey using quantitative mass spectrometry. J Biol Chem 2007; 28: 23055–23069.
Dzhekova-Stojkova S, Bogdanska J, Stojkova Z . Peroxisome proliferators: their biological and toxicological effects. Clin Chem Lab Med 2001; 39: 468–474.
Ezaki J, Komatsu M, Yokota S, Ueno T, Kominami E . Method for monitoring pexophagy in mammalian cells. Methods Enzymol 2009; 452: 215–226.
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M et al. Autophagy regulates lipid metabolism. Nature 2009; 458: 1131–1135.
Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25: 193–205.
Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino G et al. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ 2002; 9: 873–880.
Biederbick A, Kern HF, Elsässer HP . Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 1995; 66: 3–14.
Behrends C, Sowa ME, Gygi SP, Harper JW . Network organization of the human autophagy system. Nature 2010; 466: 68–76.
Hitomi K, Kitamura M, Sugimura Y . Preferred substrate sequences for transglutaminase 2: screening using a phage-displayed peptide library. Amino Acids 2009; 36: 619–624.
De Laurenzi V, Melino G . Gene disruption of tissue transglutaminase. Mol Cell Biol 2001; 21: 148–155.
Lewis MD, Pfeil J, Mueller AK . Continuous oral chloroquine as a novel route for Plasmodium prophylaxis and cure in experimental murine models. BMC Res Notes 2011; 4: 262–266.
Acknowledgements
The authors would like to thank Dr. P Mattioli from the Centre of Avanced Microscopy (CAM), Department of Biology, University of Rome Tor Vergata, for her skillful assistance in the use of the facility. This work was supported by grants from CHDI Foundation Inc. (USA), Compagnia di San Paolo, the Ministry of Health of Italy ‘Ricerca Corrente’ and ‘Ricerca Finalizzata’, and AIRC. The support of the EU grant ‘Apo-Sys’ and ‘Transpath’ Marie Curie project to MP is also acknowledged. FC is supported by grants from AIRC, Telethon and FISM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
D'Eletto, M., Farrace, M., Rossin, F. et al. Type 2 transglutaminase is involved in the autophagy-dependent clearance of ubiquitinated proteins. Cell Death Differ 19, 1228–1238 (2012). https://doi.org/10.1038/cdd.2012.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.2
Keywords
This article is cited by
-
Type 2 transglutaminase in the nucleus: the new epigenetic face of a cytoplasmic enzyme
Cellular and Molecular Life Sciences (2023)
-
Non-alcoholic fatty liver disease severity is modulated by transglutaminase type 2
Cell Death & Disease (2018)
-
The roles of PKCs in regulating autophagy
Journal of Cancer Research and Clinical Oncology (2018)
-
Polyamines: therapeutic perspectives in oxidative stress and inflammatory diseases
Amino Acids (2017)
-
Transglutaminase 2, a double face enzyme
Amino Acids (2017)

{kind=link}
{kind=link}
{kind=link}