
Abstract
Two major apoptosis pathways, the mitochondrial and death receptor pathways, are well recognized. Here we established cell lines from the fetal thymus of Apaf-1-, Caspase-9-, or Bax/Bak-deficient mice. These cell lines were resistant to apoptosis induced by DNA-damaging agents, RNA or protein synthesis inhibitors, or stress in the endoplasmic reticulum. However, they underwent efficient apoptosis when treated with kinase inhibitors such as staurosporine and H-89, indicating that these inhibitors induce a caspase-dependent apoptosis that is different from the mitochondrial pathway. CrmA, a Caspase-8 inhibitor, did not prevent staurosporine-induced apoptosis of fetal thymic cell lines, suggesting that the death receptor pathway was also not involved in this process. The staurosporine-induced cell death was inhibited by okadaic acid, a serine/threonine phosphatase inhibitor, suggesting that dephosphorylation of a proapoptotic molecule triggered the death process, or that phosphorylation of an antiapoptotic molecule could block the process. Cells of various types (fetal thymocytes, bone marrows, thymocytes, and splenocytes), but not embryonic fibroblasts, were sensitive to the noncanonical staurosporine-induced apoptosis, suggesting that the noncanonical apoptosis pathway is tissue specific.
Similar content being viewed by others


Main
Apoptosis serves to eliminate cells that are useless (e.g., interdigital cells, nonreactive lymphocytes) or harmful (e.g., tumor cells and autoreactive lymphocytes). Deregulated apoptosis causes a variety of diseases. That is, a defect in apoptosis can lead to the development of tumors and autoimmune diseases, whereas an excess of apoptosis can cause organ failure.1 Two apoptotic signaling pathways have been well characterized in mammals, the death receptor and mitochondrial pathways (also known as the extrinsic and intrinsic pathways, respectively).2, 3, 4 In the death receptor pathway, the binding of a death factor such as Fas ligand (FasL), TNF, or TNF-related apoptosis-inducing ligand (TRAIL) to its receptor triggers a signaling cascade that leads to the activation, via an adaptor, Fas-Associated protein with Death Domain (FADD), of a caspase cascade consisting of Caspase-8 and Caspase-3. In the mitochondrial pathway, cytotoxic insults upregulate or activate BH3-only proteins that activate and oligomerize Bax and Bak, and the oligomerized Bcl-2 family members Bax/Bak cause the release of cytochrome c from mitochondria into the cytosol.5 Cytochrome c then forms a complex with Apaf-1, and the cytochrome c/Apaf-1 complex, called the apoptosome, activates Caspase-9, which in turn activates Caspase-3.6, 7
The role of FADD and Caspase-8 in the death receptor-induced apoptosis pathway was confirmed by the establishment of their knockout mice.8, 9 On the other hand, conflicting reports have been published on the roles of the signaling molecules in the mitochondrial pathway. Establishing knockout mice, Cecconi et al.,10 Yoshida et al.,11 and Kuida et al.12 originally reported that Apaf-1 and caspase-9 are indispensable for genotoxic agent-induced apoptosis. However, this idea was later challenged by Marsden et al.,13 who showed that hematopoietic cells that lack Apaf-1 or Caspase-9 can undergo Bcl-2-regulated Caspase-dependent apoptosis. We recently reported that staurosporine, but not etoposide, activates Caspase-3 in embryonic day 14.5 (E14.5) fetal thymocytes in an Apaf-1-independent manner.14 Moreover, activated Caspase-3 can be detected in situ in the E14.5 Apaf-1−/− fetal thymus, suggesting that an Apaf-1-independent intrinsic apoptotic pathway exists in these cells.
Here, to characterize this Apaf-1-independent pathway, we established cell lines (immortalized fetal thymocytes (IFETs)) from the fetal thymus of wild-type and mutant mice. The Apaf-1−/− IFETs were resistant to genotoxic agent-induced apoptosis, but staurosporine caused efficient caspase-dependent apoptosis. Neither a null mutation of Caspase-9 nor double mutation of Bax/Bak abolished the staurosporine-induced apoptosis of IFETs. The expression of CrmA, a Caspase-8 inhibitor,15 also did not inhibit staurosporine-induced apoptosis, indicating that the death receptor pathway was not used in this death process. This noncanonical Apaf-1- and Caspase-8-independent apoptosis signaling pathway triggered by staurosporine was observed in lymphoid and myeloid tissues such as fetal thymocytes and bone marrow cells, but was barely detectable in embryonic fibroblasts. Together, these results suggest the existence of a new tissue-specific apoptotic pathway that is distinct from the death receptor and mitochondrial pathways.
Results
Establishment of Apaf-1-null fetal thymocyte cell line
We previously observed that Caspase-3 is activated in Apaf-1-null fetal thymus.14 To characterize the Apaf-1-independent apoptosis biochemically, it was essential to establish Apaf-1-null cell lines. Mouse fetal thymocytes were immortalized by infecting them with mouse retrovirus carrying c-myc and H-rasV12, using the RetroNectin-bound virus method. Several hundreds of transformants were obtained from the pair of thymic lobes of an Apaf-1+/+ or Apaf-1−/− E14.5 embryo. The transformants were cloned by limiting dilution, and termed IFETs. The targeted disruption of the Apaf-1 gene, or deletion of its exon 5, in Apaf-1−/− IFETs was confirmed by PCR analysis of the genomic DNA (Supplementary Figure S1A).
Incubating cell lysates with cytochrome c and dATP activates Caspase-3 in an Apaf-1-dependent manner.16 Accordingly, when extracts of Apaf-1+/+ but not Apaf-1−/− IFETs were incubated with cytochrome c and dATP, the Caspase-3 activation was observed (Supplementary Figure S1B), confirming that Apaf-1 was functionally inactivated by the deletion of its exon 5. The Apaf-1+/+ and Apaf-1−/− IFETs grew equally well, with no requirement for specific growth factors. The surface phenotype of both Apaf-1+/+ and Apaf-1−/− IFETs was Thy1.2+CD25-CD44+ (Supplementary Figure S2), although Thy1.2 was gradually downregulated after repeated passages. These results suggested that the IFETs established here were T-cell lineage cells at the early stage of development.
Apaf-1-independent Caspase-dependent cell death induced by staurosporine
Next, the wild-type and Apaf-1−/− IFETs were treated with staurosporine or etoposide. As shown in Figure 1a, the Apaf-1-null mutation blocked the etoposide-induced cell death. However, it had no effect on staurosporine-induced cell death, and >90% of staurosporine-treated Apaf-1−/− IFETs died within 12 h. A low concentration (50 μM) of caspase inhibitor (Q-VD-OPh)17 efficiently blocked the staurosporine-induced death of Apaf-1−/− as well as of wild-type IFETs (Figure 1b), although Q-VD-OPh failed to rescue the staurosporine-induced cell-cycle arrest (Figure 1c). Not only procaspase-3 but also other caspases such as procaspase-2, -6, -7, -8, and -9 were efficiently processed into its active form upon staurosporine treatment without Apaf-1 (Figure 1d), in sharp contrast to the lack of processing of procaspases by etoposide without Apaf-1. The concentration of staurosporine required to activate Caspase-3 in Apaf-1−/− IFETs seems to be higher than that required for the wild-type IFETs, but 0.1 μM staurosporine could still activate Caspase-3 in Apaf-1−/− IFETs in a time-dependent manner (Supplementary Figure S3).

Staurosporine-induced Apaf-1-independent apoptosis. (a) Apaf-1-independent cell death induced by staurosporine but not etoposide. Apaf-1+/+ (+/+) and Apaf-1−/− (−/−) IFETs were cultured with 10 μM staurosporine (STS) or 50 μM etoposide (Eto). At the indicated time, living cells were counted by Trypan blue exclusion assay, and expressed as a percentage of the initial cell number. The experiments were carried out independently for three times, and the average number was plotted with S.D. (bars). (b) Caspase-dependent Apaf-1-independent cell death induced by staurosporine. Apaf-1+/+ (+/+) and Apaf-1−/− (−/−) IFETs were pretreated with 50 μM Q-VD-OPh (Q) for 1 h, and cultured with or without 10 μM STS and 50 μM Q-VD-OPh for the indicated times. Living cells were counted by Trypan blue exclusion assay, and expressed as a percentage of the initial cell number. Experiments were performed independently for three times, and the average values were plotted with S.D. (bars). (c) No effect of caspase inhibitor on the clonogenicity of IFETs treated with staurosporine. The CFSE-labeled Apaf-1−/− IFETs were cultured in the absence or presence of Q-VD-OPh, and treated with 10 μM STS for 30 min. The cells were further cultured for 2 days in medium with or without Q-VD-OPh, stained with Annexin V/Cy5, and analyzed by FACSCalibur for CFSE and Annexin V. (d) Apaf-1-independent caspase activation by staurosporine but not etoposide. Apaf-1+/+ (WT) and Apaf-1−/− IFETs were treated with 10 μM STS for 2 h or 50 μM Eto for 6 h. Cell lysates were analyzed by western blot with anti-Caspase-3, anti-Caspase-2, anti-cleaved Caspase-6, anti-cleaved Caspase-7, anti-cleaved Caspase-8, anti-Caspase-9, anti-PARP or anti-α-tubulin Abs. Except for the western blot for PARP and Caspase-9, where the cell lysates were separated by 7.5 or 10% SDS-PAGE, respectively, the cell lysates were separated by 10–20% SDS-PAGE. Arrows indicate the pro- and activated forms of Caspase, and noncleaved (113 kDa) and cleaved PARP (85 kDa). (e) Apaf-1-independent phosphatidylserine exposure. Apaf-1+/+ (WT) and Apaf-1−/− IFETs were treated with 10 μM STS for 2 h or with 50 μM Eto for 8 h, stained with Cy5-conjugated Annexin V and PI, and analyzed by FACS. Cells with no treatment (−) were analyzed similarly, and FACS profiles are shown at left. Numbers indicate the percentage of the cells in each quadrant
During apoptotic cell death, poly(ADP)ribose polymerase (PARP) is cleaved, and phosphatidylserine is exposed to the cell surface.18, 19 Staurosporine, but not etoposide, caused PARP processing (Figure 1d) and phosphatidylserine exposure without Apaf-1 (Figure 1e), confirming that executor caspases were activated by staurosporine through an Apaf-1-independent mechanism.
No requirement for Caspase-9, Bax/Bak, or Caspase-8 in staurosporine-induced cell death
Caspase-8 and -9 are essential for executing the extrinsic and intrinsic pathways of apoptosis, respectively.8, 12 To examine the requirement for Caspase-9 in staurosporine-induced activation of Caspase-3, IFETs were established from Caspase-9−/− fetal thymocytes (Supplementary Figure S1C). As expected, etoposide could not activate Caspase-3 in the absence of Caspase-9, but staurosporine readily activated Caspase-3 without Caspase-9 (Figure 2a). Accordingly, overexpression of Caspase-9 in Caspase-9−/− IFETs had little effect on staurosporine-induced Caspase-3 activation (data not shown). IFETs were then established from fetal thymocytes of Bax−/−Bak−/− embryos20 (Supplementary Figure S1D). Staurosporine but not etoposide activated Caspase-3 in the Bax−/−Bak−/− IFETs (Figure 2a), and caused phosphatidylserine exposure. The caspase inhibitor, Q-VD-OPh, efficiently blocked staurosporine-induced phosphatidylserine exposure in Bax−/−Bak−/− IFETs, but could not rescue staurosporine-induced cell-cycle arrest (Figure 2b). The release of cytochrome c is one of the major characteristics of the mitochondrial pathway of apoptosis.21 When the wild-type and Apaf-1−/− IFETs were treated with staurosporine, the release of cytochrome c was observed, but there was no release of cytochrome c in Bax−/−Bak−/− IFETs (Figure 2c), indicating that cytochrome c was not involved in the new apoptosis pathway. Accordingly, transformation of Apaf-1−/− IFETs with human BCL-2 had no effect on staurosporine-induced Caspase-3 activation in Apaf-1−/− IFETs (Supplementary Figure S4). These results confirmed that staurosporine could activate Caspase-3 in IFETs without mitochondrial apoptotic pathway.

No requirement for the mitochondrial or death receptor apoptosis pathway for the staurosporine (STS)-induced Caspase activation. (a) Requirement of the mitochondrial pathway for etoposide-induced but not STS-induced Caspase-3 activation. Wild-type (WT), Apaf-1−/−, Caspase-9−/−, and Bax−/−Bak−/− IFETs were treated with 10 μM STS for 2 h or with 50 μM etoposide (Eto) for 6 h. The cell lysates were analyzed by western blotting with anti-Caspase-3 or anti-tubulin Abs. Arrows indicate pro- and cleaved Caspase-3. (b) No effect of caspase inhibitor on the clonogenicity of IFETs treated with STS. The CFSE-labeled Bax−/−Bak−/− IFETs were cultured in the absence or presence of Q-VD-OPh, and treated with 10 μM STS for 30 min. The cells were further cultured for 2 days in medium with or without Q-VD-OPh, stained with Annexin V/Cy5, and analyzed by FACSCalibur for CFSE and Annexin V. (c) No requirement of the cytochrome c release from mitochondria in STS-induced Caspase-3 activation. The WT, Apaf-1−/−, and Bax−/−Bak−/− IFETs were treated with 10 μM STS, permeabilized with digitonin, fixed with parafolmaldehyde, and stained with anti-cytochrome c (straight line), followed by FACS analysis. The cytochrome c staining profile before STS treatment is shown by dotted line. (d) No requirement for the death receptor pathway in STS-induced Caspase-3 activation in IFETs. Fas-Casp9−/− IFETs transformed with empty vector or CrmA were treated with 6.6 U/ml Fas ligand (FasL; left) or with 10 μM STS (right) for 2 h. The cell lysates were analyzed by western blotting with anti-Caspase-3 and anti-α-tubulin Abs. Arrows indicate pro- and cleaved Caspase-3
To examine the involvement of the extrinsic pathway of apoptosis in staurosporine-induced caspase activation, Caspase-9−/− IFETs were stably transformed with CrmA, a cowpox virus protein that inhibits Caspase-1 and -8.15 At their early developmental stage, T cells do not express Fas.22, 23 Thus, as expected, Fas was not expressed in Caspase-9−/− IFETs, and the IFETs were resistant to FasL-induced apoptosis (data not shown). When the Caspase-9−/− IFETs were transformed with Fas (Supplementary Figure S5), the FasL treatment efficiently activated Caspase-3, and this activation was inhibited by CrmA (Figure 2d). On the other hand, staurosporine activated Caspase-3 even in the presence of CrmA in Caspase-9−/− IFETs (Figure 2d). These results indicated that staurosporine causes a caspase-dependent cell death that does not use the mitochondrial or the death receptor pathway.
Various reagents activate Caspase-3 to induce apoptotic cell death. As shown in Figure 3, treatment of wild-type IFETs with 2.5 μg/ml tunicamycin, 3 μg/ml actinomycin D, 10 μg/ml cycloheximide, or γ-ray (5 Gy) irradiation efficiently activated Caspase-3, and the cells were dead within 48 h in most cases (data not shown). In contrast, IFETs lacking Apaf-1, Caspase-9, or Bax/Bak were completely resistant to apoptosis induced by these reagents (Figures 3a–c). Similarly, Caspase-3 was not activated when Bax−/−Bak−/− IFETs were treated with γ-rays (Figure 3d). These results indicated that most apoptosis-inducing reagents, such as DNA-damaging agents and protein and RNA synthesis inhibitors, required the mitochondrial pathway to activate Caspase-3.

Requirement of the mitochondrial pathway for the tunicamycin-, actinomycin D-, cycloheximide-, and γ-ray-induced caspase activation in IFETs. Wild-type, Apaf-1−/− (a–c), Caspase 9−/− (e), or Bax−/−Bak−/− (d and e) IFETs were treated with 2.5 μg/ml tunicamycin (Tun) for 6 h (a and e), 3 μg/ml actinomycin D (actD), 10 μg/ml cycloheximide (CHX) for 2 h (b and e), or 5 Gy γ-irradiation (c and d), and incubated at 37 °C for 4 h. The cell lysates were analyzed by western blotting with anti-Caspase-3 or anti-α-tubulin Abs. Arrows indicate pro- and cleaved Caspase-3
Effect of phosphatase inhibitors on staurosporine-induced cell death
Staurosporine is a multikinase inhibitor, but it also inhibits other enzymes such as ABC transporters and topoisomerase.24, 25 Thus, it is unclear whether the inhibition of a kinase or other targets led to the induction of Apaf-1-independent apoptosis. To address this question, we investigated whether phosphatase inhibitors suppressed staurosporine-induced apoptosis in Apaf-1−/− IFETs. As shown in Figure 4, okadaic acid, which is a serine/threonine phosphatase inhibitor, suppressed staurosporine-induced Caspase-3 activation (Figure 4a) and cell death in Apaf-1−/− IFETs (Figure 4b). On the other hand, sodium orthovanadate, a tyrosine phosphatase inhibitor, had only a small effect on staurosporine-induced Caspase-3 activation and cell death in Apaf-1−/− IFETs (Figure 4a and data not shown). These results suggest that staurosporine induces Apaf-1-independent apoptosis by inhibiting serine/threonine kinase(s); however, we cannot formally rule out the possibility that the phosphatase inhibitors simply enhanced an antiapoptotic pathway.

Involvement of serine/threonine kinase in the staurosporine (STS)-induced Apaf-1-independent apoptosis. (a) Effect of phosphatase inhibitors on the STS-induced Apaf-1-independent Caspase-3 activation. Apaf-1−/− IFETs were treated with 10 μM STS and 1 μM okadaic acid (OA) or 1 mM orthovanadate (VO4) for 2 h. The cell lysates were analyzed by western blotting with anti-Caspase-3 or anti-α-tubulin Abs. Arrows indicate pro- and cleaved Caspase-3. (b) Effect of OA on STS-induced Apaf-1-independent cell death. Apaf-1−/− IFETs were cultured without or with 10 μM STS or 1 μM OA, or both. At the indicated times, living cells were counted by Trypan blue exclusion assay, and are expressed as a percentage of the initial cell number. Experiments were carried out independently for three times, and the average values are plotted with S.D. (bars). (c) Apaf-1-independent Caspase-3 activation by kinase inhibitors. Apaf-1−/− IFETs were treated at 37 °C for 2 h with 10 μM STS or its derivatives (25 μM UCN-01, 50 μM GF109203X, 50 μM Ro31-6045, 50 μM Ro31-6233, 25 μM Ro31-8220, 2.5 μM K252a, 25 μM K252c, 50 μM Gö 6976, 50 μM KT5720, or 50 μM KT5823) or H-series kinase inhibitors (1 mM H-7, H-8, or H-9, or 50 μM H-89). The cell lysates were analyzed by western blotting with anti-Caspase-3 or anti-α-tubulin Abs
To confirm that inhibition of kinases induced apoptotic cell death, we examined the effect of various kinase inhibitors on apoptotic cell death. We used staurosporine analogs that had different specificities for target kinases,24, 25 H-7 and its derivatives,26 and inhibitors of MAPK and PI3K signaling pathways. Among the 10 staurosporine analogs tested, 4 (UCN-01, GF109203X, Ro31-8220, and K252a) activated Caspase-3 in Apaf-1−/− IFETs at 2.5–50 μM, whereas the others had no effect, at least at the same concentration (Figure 4c). The treatment of Apaf-1−/− IFETs with 1.0 mM H-7, H-8, or H-9 did not kill them, but the derivative of H-8, H-89, efficiently killed Apaf-1−/− IFETs and activated Caspase-3 at 50 μM. On the other hand, none of the MAPK/PI3K inhibitors tested (U0126, SP600125, SB239063, and LY294002) activated Caspase-3 in Apaf-1−/− IFETs (Supplementary Figure S6).
Cell-specific mitochondrial death pathway-independent Caspase-3 activation by staurosporine
To examine whether the new apoptosis pathway that was independent from the death receptor and mitochondrial pathways was present in other cell types, Apaf-1-null embryonic fibroblasts were prepared. As shown in Figure 5a, treatment of the primary wild-type mouse embryonic fibroblasts (MEFs) with 10 μM staurosporine generated active Caspase-3, whereas no active Caspase-3 was observed with Apaf-1−/− MEFs, indicating that MEFs carry only the canonical apoptotic pathway.

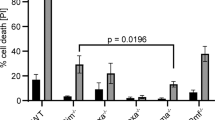
Tissue specificity of the noncanonical apoptotic pathway. Wild-type and Apaf-1−/− embryonic fibroblasts (MEFs) at passage 3 (a), and thymocytes (b and c), bone marrow cells (d), and splenocytes (e) from Baxfl/flBak−/− or Tie2Cre+Baxfl/flBak−/− mice were treated with 10 μM staurosporine (STS) for 4 h, or 50 μM etoposide (Eto) for 7 h. In (a), (c), (d), and (e), the cell lysates were analyzed by western blotting with anti-Caspase-3, anti-Bax, anti-tubulin, or anti-GAPDH Ab. Arrows indicate pro- and cleaved Caspase-3. In (b), at the indicated times, living cells were counted by Trypan blue exclusion assay, and are expressed as a percentage of the initial cell number. Experiments were carried out independently for three times, and the average values are plotted with S.D. (bars)
To examine whether the noncanonical pathway was present in adult hematopoietic and lymphoid cells, Tie2Cre+Baxfl/flBak−/− mice,27, 28, 29 in which the floxed allele of the Bax gene was deleted in hematopoietic and endothelial cells by Cre-mediated recombination (Supplementary Figure S7A), were analyzed. Western blotting analysis showed that adult thymocytes, bone marrow cells, and splenocytes from Baxfl/flBak−/− mice at the age of 7–9 weeks contained the 21-kDa Bax. In contrast, the thymocytes, bone marrow cells, and splenocytes from the Tie2Cre+Baxfl/flBak−/− mice did not show this band (Figures 5c–e), indicating that the Bax gene was fully deleted in these cells. As expected from the null mutation of Bak, the thymocytes, bone marrow cells, and splenocytes from the Baxfl/flBak−/− mice did not carry the 28-kDa Bak (Supplementary Figure S7B). Thymocytes from Baxfl/flBak−/− mice spontaneously die, and this death process was accelerated by treating with staurosporine (Figure 5b). The lack of Bax/Bak in Tie2Cre+Baxfl/flBak−/− strongly inhibited the spontaneous death, yet staurosporine killed the Tie2Cre+Baxfl/flBak−/− thymocytes.
When the thymocytes, bone marrow cells, and splenocytes from the Baxfl/flBak−/− mice were treated with staurosporine or etoposide, both reagents produced the active form of Caspase-3 (Figures 5c–e), as in wild-type fetal thymocytes. Deleting Bax and Bak in the Tie2Cre+Baxfl/flBak−/− mice completely abolished the ability of etoposide to activate Caspase-3 in these cells. On the other hand, staurosporine could still activate Caspase-3 in the absence of Bax and Bak. These results indicated that the noncanonical apoptosis pathway is present in lymphoid and myeloid cells such as thymocytes, splenocytes, and bone marrow cells, but not in fibroblasts.
Discussion
Caspase-dependent apoptotic cell death has been thought to be mediated by two pathways: the intrinsic mitochondrial pathway and the extrinsic death receptor pathway.30 Here we showed that staurosporine, a nonselective inhibitor of diverse kinases,24 could activate Caspase-3 to kill cells via two different pathways. One pathway required Apaf-1, whereas the other did not. The Apaf-1-independent caspase activation did not require Bax/Bak or Caspase-9, and was not inhibited by Bcl-2, and occurred without the release of cytochrome c from mitochondria, suggesting that the mitochondrial pathway was not involved in this death process. CrmA, an inhibitor of Caspase-8,15 also had little effect on Apaf-1-independent caspase activation, suggesting that the death receptor pathway was also not involved in this process. Recently, the activation of inflammasomes by pathogens was shown to cause Caspase-1-dependent cell death of macrophages and dendritic cells, which is called pyroptosis.31, 32 CrmA inhibits not only Caspase-8 but also Caspase-1,15 suggesting that the staurosporine-induced apoptotic pathway identified here was not pyroptosis.
The Apaf-1- and Caspase-1/8-independent noncanonical apoptotic pathway was activated by various kinase inhibitors, such as staurosporine, UCN-01, a staurosporine-derivative, and H-89. The activation of Caspase-3 by these kinase inhibitors was inhibited by okadaic acid, a phosphatase inhibitor. A simple idea is that growing cells carry a phosphatase(s) that leads to the activation of Caspase-3, and specific kinases prevent the phosphatase-induced Caspase-3 activation by keeping its target phosphorylated. Phosphatase inhibitors were previously shown to inhibit the mitochondrial and death receptor pathways,33 in which phosphatases seem to increase the death signaling that requires dephosphorylation. Therefore, it is possible that cells carry proapoptotic molecule(s) that has the potential to induce caspase-dependent apoptosis if it is not phosphorylated. Thus, a kinase(s) in growing cells may prevent the dephosphorylation of this molecule, and kinase inhibitors block this process to generate active proapoptotic molecule(s). Phosphatase inhibitors could prevent dephosphorylation of the proapoptotic molecule, or enhance antiapoptotic pathway such as the NF-κB pathway, by keeping the signaling molecules phosphorylated. Staurosporine, H-8, and their derivatives have different target specificities.24, 26, 34 A more detailed analysis of the death-inducing ability of various kinase inhibitors may help elucidate the noncanonical death signaling pathway.
Staurosporine was originally found as a molecule that induces a Bcl-2-inhibitable apoptotic death, suggesting it causes cell death via the mitochondrial pathway.35, 36 In fact, Yoshida et al.,11 by preparing Apaf-1-deficient mice, showed that staurosporine cannot activate caspase in the absence of Apaf-1. On the other hand, Stepczynska et al.37 suggested that a mitochondrial-independent pathway was responsible for staurosporine-induced apoptosis because a dominant-negative Caspase-9 cannot inhibit it. Manns et al.38 recently claimed that a noncanonical staurosporine-induced apoptosis pathway requires Caspase-9 but not Apaf-1. In this report, we showed that staurosporine could activate caspase-dependent apoptosis via two different intrinsic pathways. The first, probably present ubiquitously, is the canonical mitochondrial pathway that requires Bax/Bak, Apaf-1, and Caspase-9. The second pathway does not require the components for the mitochondrial pathway, and seems to work in a cell-type-specific manner. That is, staurosporine strongly activates the second pathway in the lymphocytes, but not embryonic fibroblasts, which apparently do not express the necessary components. This cell specificity of the noncanonical intrinsic apoptotic pathway may explain the previous controversial results on staurosporine-induced cell death, which were obtained with different cell lines and tissues.11, 37, 38
Fetal thymocytes that lacked the canonical mitochondrial apoptotic pathway, and the cell lines derived from them, efficiently underwent caspase-dependent apoptosis by staurosporine treatment, which agrees with the observation that many apoptotic cells are present in Apaf-1-null E14.5 fetal thymus.14 These apoptotic cells are observed in E14.5 but not E17.5 fetal thymus of Apaf-1-null embryos. In the E14.5 fetal thymus, most of the thymocytes are at the CD4−CD8− stage, whereas in the E17.5 or neonate thymus, most of the thymocytes are at the CD4+CD8+, CD4+CD8−, or CD4−CD8+ stage. Three major selection processes occur during thymocyte development. In one of them, β-selection, immature thymocytes that productively rearrange the gene segments of the TCRβ locus undergo proliferative expansion and mature to the CD4+CD8+ stage, whereas those failing to do so die by apoptosis.39 It is tempting to speculate that the noncanonical Apaf-1- and Caspase-1/8-independent apoptotic pathway is involved in the apoptosis accompanying β-selection. In any case, staurosporine and its derivatives (in particular, UCN-01) are under clinical trial as anticancer drugs.40, 41 Understanding how these kinase inhibitors cause apoptotic cell death will be essential for developing efficient treatment paradigms.
Materials and Methods
Mice
C57BL/6 mice were purchased from Nippon SLC (Hamamatsu, Japan). The Apaf-1−/−11 and Caspase-9−/−12 mice were maintained on a C57BL/6 background, and the Bax−/−Bak−/−20, 42 and Tie2Cre+Baxfl/flBak−/−28, 29 mice were on a mixed genetic background between C57BL/6 and 129/Sv. The mice were housed in a specific pathogen-free facility at the Kyoto University, Graduate School of Medicine, and all animal experiments were carried out in accordance with protocols approved by the Animal Care and Use Committee of the Kyoto University Graduate School of Medicine. To determine the genotype of the Apaf-1, Caspase-9, Bax, Bak, and Tie2Cre alleles, DNA from embryonic tissues or adult tail-snip tissue was analyzed using PCR. For Apaf-1, wild-type (5′-CTCAAACACCTCCTCCACAA-3′) and mutant-specific (5′-GGGCCAGCTCATTCCTC-3′) sense primers were used with a common antisense primer (5′-GTCATCTGGAAGGGCAGCGA-3′). For Caspase-9, 5′-AGGCCAGCCACCTCCAGTTCC-3′ and 5′-CAGAGATGTGTAGAGAAGCCCACT-3′ for wild-type, and 5′-CGTGCTACTTCCATTTGTCACGTC-3′ and 5′-ACACCCTTGCAAGTCAGAGGTATG-3′ for the mutant were used. For Bax, wild-type (5′-GAGCTGATCAGAACCATCATG-3′) and mutant-specific (5′-CCGCTTCCATTGCTCAGCGG-3′) sense primers and a common antisense primer (5′-GTTGACCAGAGTGGCGTAGG-3′) were used. For the floxed allele of the Bax gene, wild-type (5′-CTGGGGCGCGCGGATCCATTCCCACCG-3′) and mutant-specific (5′-TACGAAGTTATTAGGTCTGAAGAGGAG-3′) sense primers were used with a common antisense primer (5′-CCCTAGTAGTGACAAGTAGCATGGAAG-3′). For Bak, wild-type (5′-GGTGTCCACACTAGAGAACTACTC-3′) and mutant-specific (5′-CTATCAGGACATAGCGTTGG-3′) sense primers were used with a common antisense primer (5′-GAGCCATGAAGATGTTTAGC-3′). For the Tie2Cre allele, a sense primer in the Tie2 promoter region (5′-CCCTGTGCTCAGACAGAAATGAGA-3′) was used with an antisense primer in the Cre coding region (5′-CGCATAACCAGTGAAACAGCATTGC-3′).
Cells, antibodies, and reagents
MEFs were prepared from E14.5 mouse embryos as previously described,43 and cultured in DMEM containing 10% FCS (Gibco, Grand Island, NY, USA). Plat-E cells44 were cultured in DMEM containing 10% FCS, 1 μg/ml puromycin (Clontech, Palo Alto, CA, USA), and 10 μg/ml blasticidin S (Invitrogen, Carlsbad, CA, USA).
Rat anti-mouse CD16/CD32 monoclonal antibody (mAb; 2.4G2; Mouse BD Fc Block), FITC-conjugated rat anti-mouse Thy1.2 mAb (53-2.1), PE-conjugated rat anti-mouse CD25 mAb (7D4), Cy-Chrome-conjugated rat anti-mouse CD44 mAb (IM7), PE-conjugated hamster anti-mouse Fas mAb (Jo2), and mouse anti-PARP mAb (C2-10) were purchased from BD PharMingen (San Diego, CA, USA). Rabbit mAbs against Caspase-3 (8G10), GAPDH (14C10), rabbit polyclonal antibodies against cleaved caspase-6, cleaved caspase-7, and mouse cleaved caspase-8 were from Cell Signaling (Danvers, MA, USA). Rabbit anti-caspase-2 was from Abcam (Cambridge, MA, USA), and mouse anti-caspase-9 mAb (5B4) was purchased from Medical Biological Laboratories (Nagoya, Japan). Mouse anti-α-tubulin mAb (Ab-1) was from Calbiochem (San Diego, CA, USA); rabbit anti-Bax (N-20) Ab and anti-Bcl-2 (C 21) Ab were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); rabbit anti-Bak (NT) Ab was from Millipore (Temecula, CA, USA); and HRP-conjugated goat anti-rabbit and anti-mouse Ig Abs were from Dako (Copenhagen, Denmark). FITC-conjugated mouse anti-cytochrome c mAb (6H2.B4) was purchased from BioLegend (San Diego, CA, USA).
Leucine-zipper-tagged human FasL was prepared as previously described.45 Cytochrome c from bovine heart, tunicamycin, cycloheximide, etoposide, sodium orthovanadate, K252a, UCN-01, SB239063, and LY294002 were purchased from Sigma-Aldrich (St Louis, MO, USA). Actinomycin D, 4-(2–aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF), and okadaic acid were from Wako Pure Chemical (Osaka, Japan). The Caspase inhibitor, Q-VD-OPh, was from R&D Systems (Minneapolis, MN, USA). GF109203X, Ro31-8220, Ro31-6045, Ro31-6233, K252c, KT5823, and H-7 were from Enzo Life Sciences (Farmingdale, NY, USA), H-9 was from Santa Cruz, U0126 was from Promega (Madison, WI, USA), Gö 6976 was from LC Laboratories (Woburn, MA, USA), KT5720 and SP600125 were from Calbiochem, and H-89 was obtained from Cayman Chemical (Ann Arbor, MI, USA).
Plasmids
The pCX4pur, pCX4bsr, pCX4hyg, pCX4pur/H-rasV12, and pCX4bsr/c-myc retroviral vectors46 were provided by Dr. Tsuyoshi Akagi (KAN Research Institute, Kobe, Japan). The pCX4 vector was constructed by removing the drug selection marker. The coding sequence of mouse Fas47 was inserted into pCX4bsr (pCX4bsr/mFas), whereas the coding sequence of CrmA48, 49 was inserted into pCX4pur (pCX4pur/crmA). The human BCL-2-expression plasmid, pEF-BOS/BCL-2, was as previously described.50
Production of retroviruses
To produce retrovirus, Plat-E cultured in DMEM containing 10% FCS was transfected with expression vector using FuGENE 6 (Roche Applied Science, Basel, Switzerland). After culturing at 37 °C for 48 h, the supernatant was collected, passed through a 0.45-μm filter unit (Millipore, Billerica, MA, USA), and centrifuged at 6000 × g for 16 h at 4 °C.51 After centrifugation, the pellet was resuspended in a 1/10 original volume of the culture medium and stored at −80 °C.
Establishment of IFETs
Cells from fetal thymus were immortalized by infection with retroviruses carrying H-rasV12 and c-myc. That is, fetal thymocytes were cultured in the presence of IL-7,52 and then infected with retrovirus using RetroNectin kit (Takara Bio, Shiga, Japan) coupled with centrifugation, as recommended by the supplier. In brief, the wells of a 96-well plate (flat-bottom, BD Falcon) were coated with 6 μg of RetroNectin at 4 °C overnight, and then incubated at room temperature for 30 min in PBS containing 2% BSA. The retroviruses were added to the wells, and the plate was centrifuged at 2000 × g for 2 h at room temperature, and washed with PBS. Primary E14.5 mouse fetal thymocytes were suspended in culture medium (DMEM containing 10% FCS, 1 × NEAA (non-essential amino acids), 50 μM 2-mercaptoethanol, and 10 mM HEPES-NaOH (pH 7.4)) supplemented with 5 ng/ml mouse IL-7 (PeproTech, Rocky Hill, NJ, USA) and added to the wells. The cells were attached to the wells by centrifuging at 400 × g for 5 min, and the plate was incubated for 3 days at 37 °C. E14.5 Apaf-1+/+ and Apaf-1−/− fetal thymocytes were infected with retroviruses from pCX4pur/H-rasV12 and pCX4bsr/c-myc. Other thymocytes were infected with retroviruses from pCX4/H-rasV12 and pCX4/c-myc. A single clone of each genotype was obtained by limiting dilution.
Transfection of IFETs
To express mouse Fas and CrmA, 1 × 105 IFETs in 12-well flat-bottom plates were suspended in culture medium containing retrovirus and 10 μg/ml polybrene (Sigma-Aldrich), centrifuged at 1000 × g for 1 h at room temperature,53, 54 and cultured at 37 °C for 24 h. Puromycin or blasticidin was added to the culture at a final concentration of 5 or 50 μg/ml, and the cells were further incubated for 7 or 10 days, respectively. The BCL-2 expression plasmid, pEF-BOS/BCL-2, was introduced into IFETs by electroporation together with hygromysin-resistant gene pCX4hyg, and the stable transformants were selected by culturing in the presence of 4 mg/ml hygromycin B (Wako Pure Chemical).
FACS analysis
To examine the expression of surface antigens, 1 × 105 IFETs in 96-well round-bottom plates were centrifuged at 300 × g for 3 min at 4 °C, washed with FACS staining buffer (2% FCS and 0.02% NaN3 in PBS), pretreated with Fc block, stained with fluorescent dye-conjugated antibodies, and analyzed by FACSCalibur (BD Biosciences, San Jose, CA, USA).
To detect phosphatidylserine exposed on apoptotic cells, 6.25 × 104 cells were incubated at room temperature for 15 min with 500-fold diluted Cy5-conjugated Annexin V (BioVision, Mountain View, CA, USA) in 0.5 ml of Annexin V staining buffer (10 mM HEPES-NaOH (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2). Propidium iodide (PI) was added to a final concentration of 2.5 μg/ml, and the samples were incubated at room temperature for 5 min, and analyzed by FACSCalibur.55
Cell-free assay for Caspase-3 activation
The cytochrome c-induced activation of Caspase-3 was assayed essentially as described.56 In brief, IFETs were harvested by centrifugation at 400 × g for 5 min at 4 °C, washed with extraction buffer (50 mM PIPES-KOH (pH 7.0), 20 mM KCl, 5 mM EGTA, 1 mM DTT, and 10 mM AEBSF), and suspended in 0.8 volumes of the extraction buffer. After incubation on ice for 20–30 min, the cells were disrupted in a Dounce homogenizer (Wheaton, Millville, NJ, USA) with 100 strokes of a tight pestle. The cell lysates were subjected to centrifugation at 10 000 × g for 10 min at 4 °C, and the supernatant was further centrifuged at 100 000 × g for 1 h at 4 °C to obtain the S-100 fraction. Protein (100 μg) of the S-100 fraction was incubated at 37 °C for 30 min with 200 μM dATP and 4 μM cytochrome c in 25 μl of the extraction buffer. The reaction was stopped by placing the reaction mixture on ice, and the sample was mixed with an equal volume of 2 × SDS sample buffer (125 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 0.02% BPB, and 10% 2-mercaptoethanol) for western blotting.
Assay for apoptosis
To induce apoptosis, IFETs, primary thymocytes and splenocytes, bone marrow cells treated with erythrocyte lysis buffer, and MEFs at passage 3 were suspended at 1 × 106, 1 × 107, 5 × 106, and 5 × 104 cells per ml in culture medium, respectively, and incubated at 37 °C with 10 μM staurosporine and 50 μM etoposide. To induce apoptosis by γ-rays, the cells were irradiated with 5 Gy using a 137Cs gamma-irradiator (dose rate of 0.744 Gy/min in a Gammacell 40 Exactor, Nordion International, Kanata, Ontario, Canada), and incubated at 37 °C. Viable cells were counted by Trypan blue exclusion assay.
Western blot analysis
Cells were harvested by centrifugation at 1100 × g for 5 min at 4 °C, washed with PBS, suspended at 1.0–10 × 107 cells/ml in RIPA buffer (50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP40, 0.1% SDS, and 0.5% sodium deoxycholate) containing a protease inhibitor cocktail (cOmplete, Mini, Roche), and incubated on ice for 15 min. Samples were centrifuged at 13 000 × g for 15 min at 4 °C. The protein concentration of the supernatant was adjusted to 0.5 mg/ml with RIPA buffer, and the sample was mixed with an equal volume of 2 × SDS sample buffer. After incubating at 98 °C for 5 min, the proteins were separated by 10–20% gradient SDS-PAGE if not specified, and transferred to PVDF membranes (pore size: 0.22 or 0.45 μm, Millipore). After incubation at room temperature for 1 h with blocking buffer (TBST (Tris-buffered saline containing 0.05% Tween 20) supplemented with 5% non-fat dry milk), the membranes were stained with primary antibodies in blocking buffer or solution 1 (Can Get Signal Immunoreaction Enhancer Solution; Toyobo, Osaka, Japan) at room temperature for 1 h or at 4 °C overnight. After being washed with TBST, the membranes were incubated at room temperature for 1 h with HRP-conjugated secondary antibodies in blocking buffer or solution 2. The proteins recognized by the antibody were then visualized by a chemiluminescence reaction (Immobilon Western, Millipore) and detected by the LAS-4000 system (Fujifilm, Tokyo, Japan).
Clonogenic assay
After washing with PBS containing 0.1% BSA, IFETs were resuspended at a final concentration of 1 × 106 cells/ml in pre-warmed PBS containing 0.1% BSA and 10 μM CFSE (Molecular Probes, Eugene, OR, USA), and incubated at 37 oC for 10 min. The staining reaction was stopped by addition of 5 volumes of ice-cold IFET culture medium, and incubated on ice for 5 min. After washing twice with ice-cold culture medium, the CFSE-stained cells were pretreated with 50 μM Q-VD-OPh for 1 h in culture medium, and further treated at 37 °C for 30 min with 10 μM staurosporine in the presence of 50 μM Q-VD-OPh. The cells were then washed with ice-cold culture medium, and cultured at 37 °C for 2 days in the culture medium containing 50 μM Q-VD-OPh. Then, the cells were stained at room temperature for 15 min with 1 : 1000 diluted Cy5/Annexin V in Annexin V staining buffer, and were analyzed by FACSCalibur.
Cytochrome c release assay
The apoptotic cytochrome c release was assayed as described previously.57 In brief, 1 × 105 IFETs were treated at 37 °C for 2 h with 10 μM staurosporine, and treated for 5 min on ice with 50 μg/ml digitonin in 100 μl of PBS containing 100 mM KCl until >95% cells were permeabilized as assessed by Trypan blue exclusion. IFETs were washed with FACS staining buffer, fixed for 20 min at room temperature with 4% paraformaldehyde in PBS, washed three times, and incubated at room temperature for 1 h in blocking buffer (PBS containing 3% BSA and 0.05% saponin). The cells were then incubated for 1 h at room temperature in 1 : 200 diluted FITC-conjugated anti-cytochrome c mAb, washed, and analyzed by FACSCalibur.
Abbreviations
- E14.5:
-
embryonic day 14.5
- FADD:
-
Fas-associated protein with death domain
- FasL:
-
Fas ligand
- IFET:
-
immortalized fetal thymocyte
- MEF:
-
mouse embryonic fibroblast
- PARP:
-
poly(ADP)ribose polymerase
- TRAIL:
-
TNF-related apoptosis-inducing ligand
References
Vaux DL, Korsmeyer SJ . Cell death in development. Cell 1999; 96: 245–254.
Strasser A, Jost PJ, Nagata S . The many roles of FAS receptor signaling in the immune system. Immunity 2009; 30: 180–192.
Krammer PH . CD95’s deadly mission in the immune system. Nature 2000; 407: 789–795.
Nagata S . Apoptosis by death factor. Cell 1997; 88: 355–365.
Westphal D, Dewson G, Czabotar PE, Kluck RM . Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta 2011; 1813: 521–531.
Ow YP, Green DR, Hao Z, Mak TW . Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol 2008; 9: 532–542.
Riedl SJ, Salvesen GS . The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 2007; 8: 405–413.
Varfolomeev EE, Schuchmann M, Luria V, Chiannikulchai N, Beckmann JS, Mett IL et al. Target disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998; 9: 267–276.
Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998; 279: 1954–1958.
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P . Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998; 94: 727–737.
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998; 94: 739–750.
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998; 94: 325–337.
Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 2002; 419: 634–637.
Nagasaka A, Kawane K, Yoshida H, Nagata S . Apaf-1-independent programmed cell death in mouse development. Cell Death Differ 2010; 17: 931–941.
Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS . Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem 1997; 272: 7797–7800.
Zou H, Li Y, Liu X, Wang X . An APAF-1.Cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 1999; 274: 11549–11556.
Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL . Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 2003; 8: 345–352.
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM . Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 1992; 148: 2207–2216.
Soldani C, Scovassi AI . Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis 2002; 7: 321–328.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399.
Youle R, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Ogasawara J, Suda T, Nagata S . Selective apoptosis of CD4 CD8 thymocytes by the anti-Fas antibody. J Exp Med 1995; 181: 485–491.
Andjelić S, Drappa J, Lacy E, Elkon KB, Nikolić-Zugić J . The onset of Fas expression parallels the acquisition of CD8 and CD4 in fetal and adult alpha beta thymocytes. Int Immunol 1994; 6: 73–79.
Nakano H, Omura S . Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J Antibiot (Tokyo) 2009; 62: 17–26.
Gani OA, Engh RA . Protein kinase inhibition of clinically important staurosporine analogues. Nat Prod Rep 2010; 27: 489–498.
Hidaka H, Kobayashi R . Pharmacology of protein kinase inhibitors. Annu Rev Pharmacol Toxicol 1992; 32: 377–397.
Schlaeger TM, Mikkola HKA, Gekas C, Helgadottir HB, Orkin SH . Tie2Cre-mediated gene ablation defines the stem-cell leukemia gene (SCL/tal1)-dependent window during hematopoietic stem-cell development. Blood 2005; 105: 3871–3874.
Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M . Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 2001; 230: 230–242.
Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ . Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci USA 2005; 102: 11272–11277.
Strasser A, O’Connor L, Dixit VM . Apoptosis signaling. Annu Rev Biochem 2000; 69: 217–245.
Miao EA, Rajan JV, Aderem A . Caspase-1-induced pyroptotic cell death. Immunol Rev 2011; 243: 206–214.
Brodsky IE, Medzhitov R . Pyroptosis: macrophage suicide exposes hidden invaders. Curr Biol 2011; 21: R72–R75.
Chatfield K, Eastman A . Inhibitors of protein phosphatases 1 and 2A differentially prevent intrinsic and extrinsic apoptosis pathways. Biochem Biophys Res Commun 2004; 323: 1313–1320.
Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005; 23: 329–336.
Jacobsen MD, Weil M, Raff MC . Role of Ced-3/ICE-family proteases in staurosporine-induced programmed cell death. J Cell Biol 1996; 133: 1041–1051.
Jacobson MD, Burne JF, Raff MC . Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J 1994; 13: 1899–1910.
Stepczynska A, Lauber K, Engels IH, Janssen O, Kabelitz D, Wesselborg S et al. Staurosporine and conventional anticancer drugs induce overlapping, yet distinct pathways of apoptosis and caspase activation. Oncogene 2001; 20: 1193–1202.
Manns J, Daubrawa M, Driessen S, Paasch F, Hoffmann N, Löffler A et al. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: activation of caspase-9 in the absence of Apaf-1. FASEB J 2011; 25: 3250–3261.
Wiest DL, Berger MA, Carleton M . Control of early thymocyte development by the pre-T cell receptor complex: a receptor without a ligand? Semin Immunol 1999; 11: 251–262.
Lapenna S, Giordano A . Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 2009; 8: 547–566.
Fuse E, Kuwabara T, Sparreboom A, Sausville EA, Figg WD . Review of UCN-01 development: a lesson in the importance of clinical pharmacology. J Clin Pharmacol 2005; 45: 394–403.
Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ . Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 1995; 270: 96–99.
Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S . Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 2005; 202: 1333–1339.
Morita S, Kojima T, Kitamura T . Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 2000; 7: 1063–1066.
Shiraishi T, Suzuyama K, Okamoto H, Mineta T, Tabuchi K, Nakayama K et al. Increased cytotoxicity of soluble Fas ligand by fusing isoleucine zipper motif. Biochem Biophys Res Commun 2004; 322: 197–202.
Akagi T, Sasai K, Hanafusa H . Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc Natl Acad Sci USA 2003; 100: 13567–13572.
Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA et al. The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol 1992; 148: 1274–1279.
Pickup DJ, Ink BS, Hu W, Ray CA, Joklik WK . Hemorrhage in lesions caused by cowpox virus is induced by a viral protein that is related to plasma protein inhibitors of serine proteases. Proc Natl Acad Sci USA 1986; 83: 7698–7702.
Enari M, Hug H, Nagata S . Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature 1995; 375: 78–81.
Itoh N, Tsujimoto Y, Nagata S . Effect of bcl-2 on Fas antigen-mediated cell death. J Immunol 1993; 151: 621–627.
Bowles NE, Eisensmith RC, Mohuiddin R, Pyron M, Woo SL . A simple and efficient method for the concentration and purification of recombinant retrovirus for increased hepatocyte transduction in vivo. Hum Gene Ther 1996; 7: 1735–1742.
Watson JD, Morrissey PJ, Namen AE, Conlon PJ, Widmer MB . Effect of IL-7 on the growth of fetal thymocytes in culture. J Immunol 1989; 143: 1215–1222.
Bunnell B, Muul L, Donahue R, Blaese R, Morgan R . High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Sci USA 1995; 92: 7739–7743.
Travers H, Anderson G, Gentle D, Jenkinson E, Girdlestone J . Protocols for high efficiency, stage-specific retroviral transduction of murine fetal thymocytes and thymic epithelial cells. J Immunol Methods 2001; 253: 209–222.
Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C . A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995; 184: 39–51.
Genini D, Budihardjo I, Plunkett W, Wang X, Carrera CJ, Cottam HB et al. Nucleotide requirements for the in vitro activation of the apoptosis protein-activating factor-1-mediated caspase pathway. J Biol Chem 2000; 275: 29–34.
Waterhouse NJ, Trapani JA . A new quantitative assay for cytochrome c release in apoptotic cells. Cell Death Differ 2003; 10: 853–855.
Acknowledgements
We thank Dr. Tsuyoshi Akagi (KAN Research Institute, Kobe, Japan) for the pCX4 series vectors, Dr. Shin Yonehara (Graduate School of Biostudies, Kyoto University) for the Caspase-9−/− mice in a C57/B6 background, and Dr. Osamu Takeuchi (Virus Institute, Kyoto University) for the Bax−/−Bak−/− and Baxfl/flBak−/− mice, and the Tie2-Cre transgenic mice. We are grateful to Drs. Masamichi Ishiai and Minoru Tanaka (Radiation Biology Center, Kyoto University) for help with the γ-irradiation. We thank Ms. Makiko Fujii for secretarial assistance. This work was supported in part by Grants-in-Aid for Specially Promoted Research from the Ministry of Education, Science, Sports, and Culture in Japan. TI is a research fellow of the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Vaux
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Imao, T., Nagata, S. Apaf-1- and Caspase-8-independent apoptosis. Cell Death Differ 20, 343–352 (2013). https://doi.org/10.1038/cdd.2012.149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.149
