
Abstract
Hypoxia-inducible factor (HIF) 1α and HIF2α and the inhibitor of apoptosis survivin represent prominent markers of many human cancers. They are also widely expressed in various embryonic tissues, including the central nervous system; however, little is known about their functions in embryos. Here, we show that zebrafish HIF2α protects neural progenitor cells and neural differentiation processes by upregulating the survivin orthologues birc5a and birc5b during embryogenesis. Morpholino-mediated knockdown of hif2α reduced the transcription of birc5a and birc5b, induced p53-independent apoptosis and abrogated neural cell differentiation. Depletion of birc5a and birc5b recaptured the neural development defects that were observed in the hif2α morphants. The phenotypes induced by HIF2α depletion were largely rescued by ectopic birc5a and birc5b mRNAs, indicating that Birc5a and Birc5b act downstream of HIF2α. Chromatin immunoprecipitation assay revealed that HIF2α binds to birc5a and birc5b promoters directly to modulate their transcriptions. Knockdown of hif2α, birc5a or birc5b reduced the expression of the cdk inhibitors p27/cdkn1b and p57/cdkn1c and increased ccnd1/cyclin D1 transcription in the surviving neural progenitor cells. The reduction in elavl3/HuC expression and enhanced pcna, nestin, ascl1b and sox3 expression indicate that the surviving neural progenitor cells in hif2α morphants maintain a high proliferation rate without terminally differentiating. We propose that a subset of developmental defects attributed to HIF2α depletion is due in part to the loss of survivin activity.
Similar content being viewed by others


Main
Hypoxia-inducible factors (HIFs), including HIF1, HIF2 and HIF3, are known to function in oxygen homeostasis by regulating the genes responsible for glucose uptake and metabolism, erythropoiesis, angiogenesis, apoptosis and cell proliferation.1 The HIFs are heterodimeric basic-helix-loop-helix-PAS transcription factors that consist of an oxygen-regulated α subunit and a constitutively expressed β subunit, which is also known as ARNT.1 In normoxic conditions, both HIF1α and HIF2α are targeted for proteosomal degradation by prolyl hydroxylase and the von Hippel–Lindau (VHL) E3 ligase complex.2 When cells are subjected to hypoxia, the HIF-α factors are stabilized and in turn associate with ARNT and activate target genes.2 HIF3α lacks a conventional C-terminal transactivation domain and it is postulated to act as a negative regulator of hypoxia-inducible gene expression.3
Despite their oxygen homeostatic functions in adult tissues, HIF-related pathways also have critical functions in embryos. Constitutive depletion of the mouse HIF1α gene (HIF1α−/−) results in developmental arrest and embryonic lethality, which is characterized by neural tube defects, cardiovascular malformations, dysregulated erythropoiesis signaling, and marked cell death within the cephalic mesenchyme.4, 5, 6 HIF2α null (HIF2α−/−) embryos died as a result of inadequate blood vessel fusion, remodeling and impaired fetal lung maturation.7, 8 Inhibition of HIF2α expression also enhanced the generation of reactive oxygen species (ROS) and reduced transcription of primary anti-oxidant enzymes (AOEs), which in turn caused a syndrome of multiple-organ pathology.9 Neural cell-specific depletion of HIF1α resulted in hydrocephalus accompanied by an increase in neuron cell apoptosis and vascular regression in the telencephalon of mutant mouse embryos.10
Depending on the severity of hypoxia, hypoxic signals may induce different responses during cell death. For the pro-apoptotic pathway, HIF1α conspires with p53 and/or BNIP3 to promote apoptosis.11, 12 However, hypoxia can also induce an anti-apoptotic response by increasing the expression of the anti-apoptotic protein IAP2 and suppressing the expression of the pro-apoptotic protein Bax through a HIF1α-independent mechanism.13 A recent study has shown that HIF2α may be involved in the anti-apoptotic properties of tumor cells. Inhibition of HIF2α promoted p53 activity and induced tumor cell death by disturbing cellular redox homeostasis and promoting the accumulation of ROS.14
Survivin (Birc5) is the smallest member of the inhibitor of apoptosis proteins (IAPs) and contains a single baculovirus IAP repeat (BIR) domain and an extended C-terminal α-helical coiled-coil domain.15 In addition to inhibiting apoptosis, survivin also has important functions in cell mitosis and cell-cycle progression.16 The mammalian survivin protein is widely expressed in embryonic cells, especially in neural progenitor cells, but it is barely detectable in quiescent adult cells.17 During brain development, survivin is highly expressed in neural progenitor cells. Targeted deletion of survivin in neural precursor cells leads to massive apoptosis in the central nervous system (CNS) due to elevated caspase-3 and caspase-9 activities.18 Interestingly, survivin is widely expressed in all kinds of malignant tumors, making it a potent target for cancer therapy.15, 19
There are multiple HIFα factors, including HIF1α, HIF2α and HIF3α, in zebrafish that are widely expressed during development.20, 21 Concurrent knockdown of these three factors leads to neural cell death and abrogates neuronal development, indicating that these HIF-α factors have critical roles in neural cell survival and differentiation.20 Nevertheless, the authentic HIF-α factor responsible for the fates of CNS neuronal progenitor cells (NPCs) remains to be elucidated. Here, we demonstrate that of the three HIF-α factors, HIF2α has a major role in maintaining cell survival and promotes neural progenitor cell differentiation. HIF2α depletion caused massive cell death and abrogated neural cell differentiation due to aberrant expression of the survivin homologs (birc5a and birc5b). The surviving NPCs remained in the cell cycle. The defects that appeared in hif2α morphant embryos were rescued by ectopic injection of the birc5a or birc5b mRNA, suggesting that survivins act downstream of HIF2α to protect neural progenitor cells and promote neural differentiation. Chromatin immunoprecipitation assay revealed that HIF2α binds to both birc5a and birc5b promoters directly to modulate their transcriptions.
Results
HIF2 α knockdown induces p53-independent apoptosis. There are multiple HIF-α factors, including HIF1α, HIF2α and HIF3α, in zebrafish that are widely expressed during development.20, 21 Concurrent knockdown of these three factors leads to neural cell death and abrogates neuronal development.20 To clarify the particular HIF α factor that determines the fates of zebrafish CNS NPCs, we analyzed apoptotic events in individual hifα morphant embryos. We found that knockdown of hif2α by either of two distinct anti-sense morpholinos resulted in massive apoptosis at the 24- and 48-h post-fertilization (h.p.f.) stage (Figures 1a–c, g–h and t). Conversely, knockdown of hif1α and hif3α either individually or concurrently did not increase the number of apoptotic cells (Figures 1d–f, i and t), indicating that HIF2α has a unique function in protecting embryonic cells against apoptosis.

HIF2α protects CNS neural progenitors from cell death. (a–f) Lateral views of acridine orange (AO) staining of wild-type (WT; a), hif2α translation-blocking MO (hif2α ATG-MO; b), hif2α SPL-MO (c), hif1α ATG-MO (d), hif3α ATG-MO (e) and hif1α, hif3α dual ATG-MO (f) 24 h.p.f. embryos. The apoptotic cells are labeled with white spots. (g–i) Transverse brain sections with TUNEL (bright green) and DAPI (blue) staining in WT (g), hif2α ATG-MO (h) and hif1α, hif3α dual ATG-MO (i) 48 h.p.f. embryos. (j and k) Lateral views of acridine orange (AO) staining of WT (j), hif2α ATG-MO (k) 12 h.p.f. embryos. (l and m) Lateral views of pcna expression in WT (l) and hif2α ATG-MO (m) 12 h.p.f. embryos. (n and o) Apoptosis analysis of p53 ATG-MO (n) and p53, hif2α dual ATG-MO (o) 24 h.p.f. embryos by AO staining. (p and q) Apoptosis analyses of control (p) and hif2α ATG-MO (q) 42 h.p.f. p53M214K mutant embryos by AO staining. (r and s) Colocalization of TUNEL signaling (in green) and nes expression (in red) in WT (r) and hif2α ATG-MO (s) 56 h.p.f. embryos. nes was stained by fluorescence in situ hybridization. The TUNEL-positive, nes-positive cells (in yellow) represent the apoptotic NPCs. (t) Percentages of normal, moderate and severe apoptosis in the WT control (n=112), hif1α ATG-MO (n=72), hif2α ATG-MO (n=114), hif2α SPL-MO (n=66), hif3α ATG-MO (n=50) and hif1α, hif3α dual ATG-MO (n=137) embryos
In addition to the massive apoptosis, a morphological abnormality with small and acellular head was observed in hif2α morphants at the 48-h.p.f. stage (Figure 1h). During early stage of development, massive apoptosis was elicited by hif2α morpholino at the 12-h.p.f. stage before any morphological defect observed (Figures 1j and k). Moreover, the pcna expression in the anterior proliferative region was diminished at the 12-h.p.f. stage, possibly due to the considerable cell death (Figures 1l and m). It suggests that the gross developmental abnormalities observed in hif2α morphants is possibly caused by extensive cell death occurred in the early stage of development.
A similar anti-apoptotic function has been observed for the human HIF2α protein. siRNA-mediated knockdown of HIF2α promoted p53 activity in human tumor cells and induced cell death by disturbing the cellular redox homeostasis and prompting the accumulation of ROS.14 To examine whether the hif2α morpholino-induced apoptosis in zebrafish embryos is mediated by p53 activity, we concurrently knocked down p53 and hif2α using translational morpholinos. Our results show that the depletion of p53 expression did not block the massive apoptosis induced by treatment with hif2α morpholinos (Figures 1n and o). Similarly, severe apoptosis was observed in p53M214K mutant embryos after injection with a hif2α translation-blocking MO (Figures 1p and q), indicating that the HIF2α depletion-induced apoptosis is not an off-target effect of the morpholino and that it is mediated through a p53-independent pathway.
To verify whether the apoptotic cells observed in hif2α morphants are undifferentiated NPCs, embryos were first in situ hybridized with fluorescence nestin (nes) RNA probe followed by TUNEL assay. We found that most of apoptotic cells were colocalized with nes-positive cells (as labeled yellow in Figures 1r and s), indicating that the apoptotic cells in hif2α morphants are indeed NPCs.
Knockdown of hif2 α abrogates CNS differentiation. We next examined the fate of the surviving NPCs in the hifα morphant embryos using the terminal differentiation marker elavl3. Knockdown of hif2α caused a dramatic reduction in elavl3 transcription (Figures 2b, c and m) compared with wild-type embryos (Figure 2a) or the other two types of hifα morphants (Figures 2d–f and m), indicating that loss of hif2α expression not only stimulated NPC apoptosis but also abrogated the differentiation of these cells. The loss of neural differentiation in hif2α morphants was confirmed by the lack of HuC/D expression (Figure 2h) and acetylated α-tubulin (Figure 2k). Consistent with this observation, the hif2α morphants had higher transcript levels of the neural progenitor markers, such as nes, sox3 and achaete-scute complex-like 1b (ascl1b), compared with control embryos in the late stages of development (Figures 3a–f), suggesting that the NPCs are retained in the undifferentiation stage. The high level of PCNA expression in CNS suggests that the undifferentiated NPCs in the hif2α morphants maintain a high level of proliferation (Figures 3h and j). Moreover, the reduction in the expression of the cdk inhibitors cdkn1b (Figures 4a, b and g) and cdkn1c in the CNS region (Figures 4c, d and h) was accompanied by an increase in ccnd1 transcription (Figures 4e, f and i), indicating that the NPCs in the hif2α morphants did not exit normally from the cell cycle. It was further supported by double fluorescent in situ hybridization of nes and ccnd1 expression (Figures 4j–o). The colocalization of these two gene expression in the CNS of hif2α morphants (yellow in Figure 4o) suggesting that most of NPCs in these hif2α morphants were hyperproliferating cells.

Knockdown of hif2α impairs CNS development. (a–f) Lateral views of elavl3 expression in WT (a), hif2α ATG-MO (b), hif2α SPL-MO (c), hif1α ATG-MO (d), hif3α ATG-MO (e) and hif1α, hif3α dual ATG-MO (f) 24 h.p.f. embryos. elavl3 expression in the CNS was decreased greatly under the hif2α ATG-MO and hif2α SPL-MO treatments, indicating that these NPCs were not terminally differentiated. (g–i) Immunofluorescent staining of HuC/HuD (in red) in transverse brain sections of WT (g), hif2α ATG-MO (h) and hif1α, hif3α dual ATG-MO (i) 48 h.p.f. embryos. Nuclei are labeled by DAPI staining (in blue). Consistent with the mRNA transcriptional activity, knockdown of hif2α eliminated HuC expression, indicating a defect in neural differentiation. Knockdown of hif2α also caused morphological abnormality with small head. (j–l) Dorsal views of whole-mount immunostained images of acetylated α-tubulin in WT (j), hif2α ATG-MO (k) and hif1α/hif3α ATG-MO (l) 24 h.p.f. embryos. Axons were visualized by antibody staining for acetylated α-tubulin. The neural development defect following hif2α MO treatment was reflected by the loss of axon growth. (m) Percentages of normal, moderate and severe loss of elavl3 expression in the WT control (n=50), hif1α ATG-MO (n=54), hif2α ATG-MO (n=69), hif2α SPL-MO (n=23), hif3α ATG-MO (n=25) and (n=64) embryos. Knockdown of hif2α with either translation-blocking or splicing-blocking MOs led to a severe loss of elavl3 transcription, indicating impairment in neural differentiation

The surviving neural progenitor cells in hif2α morphants continue to proliferate without terminally differentiating. (a–f) Lateral views of nes (a and b), sox3 (c and d) and ascl1b (e and f) expression in WT (a, c and e) and hif2α ATG-MO (b, d and f) 48 h.p.f. embryos. nes, sox3 and ascl1b were highly expressed in the NPCs but not in mature neural cells. The high level of nes, sox3 and ascl1b transcriptions in hif2α morphants suggests that the NPCs did not differentiate into mature neural cells. (g and h) Lateral views of pcna transcription in WT (g) and hif2α ATG-MO (h) 48 h.p.f. embryos. (i and j) Immunofluorescent staining of PCNA (in red) in transverse brain sections of WT (i) and hif2α ATG-MO (j) 48 h.p.f. embryos. Nuclei are labeled by DAPI staining (in blue). The increased pcna expression in the CNS of hif2α MO embryos indicates that the NPCs were proliferating

The surviving neural progenitor cells in hif2α morphants do not exit normally from the cell cycle. (a–f) Lateral views of cdkn1b (a and b), cdkn1c (c and d) and ccnd1c (e and f) expression in WT (a, c and e) and hif2α ATG-MO (b, d and f) 48 h.p.f. embryos. (g–i) Quantitative RT-PCR of the cdkn1b (g), cdkn1c (h) and ccnd1 (i) genes in WT, hif2α ATG-MO and hif1α, hif3α dual ATG-MO 24 h.p.f. embryos. Decreased cdkn1b and cdkn1c expression and increased ccnd1 expression in the CNS of hif2α morphants suggest that the NPCs failed to exit from the cell cycle. (j–o) Double fluorescent in situ hybridizations of nes (red; j and k) and ccnd1 (green; l and m) in WT (j, l and n) and hif2α ATG-MO (k, m and o) 48 h.p.f. embryos. Merged images (n and o) are shown. The colocalization of nes and ccnd1 (in yellow) expressions in hif2α morphants indicates most of surviving NPCs are hyperproliferating cells
Survivins function downstream of HIF2 α to protect neural progenitor cells and promote neural differentiation. Survivin is the smallest member of the IAP family and has multiple functions in cell survival, mitosis and proliferation. Knockdown of the zebrafish survivin orthologues birc5a and birc5b causes profound neuro-developmental, hematopoietic, cardiogenic, vasculogenic and angiogenic defects.22 Together, these results suggest that survivin functions in embryonic development. In various human cancer cells, survivin/BIRC5 transcription is upregulated as a result of increases in HIF1α.23 This raises the possibility that the survivin orthologues in zebrafish are also affected by one of the HIF factors. Here, we analyzed the transcriptional activities of the survivin orthologues birc5a and birc5b in various hifα morphant embryos. In wild-type embryos, the survivin orthologues birc5a and birc5b and hif2α were all transcribed intensively in the CNS region (Figures 5a–c). Depletion of HIF2α resulted in a reduction in the transcription of both birc5a and birc5b (Figures 5d, e, h and i), indicating that hif2α correlates with survivin expression. Conversely, concurrent depletion of hif1α and hif3α did not affect the transcription of either birc5a or birc5b (Figures 5f–i). This suggests that zebrafish survivin orthologues are affected specifically by HIF2α but not by other HIF-α factors. To determine whether the survivin deficiency accounts for the HIF2α depletion-induced NPC apoptosis and aberrant neural differentiation, we examined NPC apoptosis and neural differentiation in birc5a and birc5b morphant embryos. Knockdown of either birc5a or birc5b caused considerable increases in apoptosis and morphological abnormality, which were similar to that observed in hif2α morphant embryos (Figures 6a, b, g, h, 7a–c and 8). Additionally, the birc5a and birc5b morphants exhibited much lower levels of elavl3 transcription compared with WT controls (Figures 6c, e and i–l). Likewise, birc5a and birc5b morphants exhibited higher levels of nes and pcna transcription compared with controls (Figures 9a–i). Moreover, the birc5a and birc5b morphant embryos had lower levels of cdkn1b and cdkn1c transcription and higher levels of ccnd1 transcription than control embryos (Figures 9j–r). Together, these observations indicate that knockdown of either birc5a or birc5b results in the induction of NPC apoptosis and the inhibition of cell-cycle exit in the NPCs, which is similar to the phenotype observed in the hif2α morphant embryos. Moreover, ectopic treatment with the birc5a or birc5b mRNA not only successfully prevented hif2α MO-induced apoptosis and morphological abnormality (Figures 6m, n, 7d and 8), but also restored elavl3 expression to a nearly normal level (Figures 6o–r), indicating that the survivin orthologues indeed rescue the impairment in NPC development that is caused by HIF2α deficiency.

Knockdown of hif2α reduces the expression of the survivin orthologues. (a–c) Lateral views of hif2α (a), birc5a (b) and birc5b (c) transcription in 24 h.p.f. WT embryos. Expression of all three transcripts overlapped in the CNS region. (d–g) Lateral views of birc5a (d and f) and birc5b (e and g) expression in hif2α ATG-MO (d and e) and hif1α, hif3α dual ATG-MO (f and g) 24 h.p.f. embryos. The expression levels of both birc5a and birc5b were reduced in hif2α morphants. (h and i) Quantitative RT-PCR of the birc5a (h) and birc5b (i) genes in WT, hif2α ATG-MO and hif1α, hif3α dual ATG-MO 24-h.p.f. stage embryos. Expression was normalized to β-actin. Knockdown of hif2α caused a reduction in the transcription of birc5a and birc5b

Survivin orthologues function downstream of HIF2α to control CNS development. (a and b) Transverse brain sections with TUNEL staining in 48 h.p.f. wild-type (WT; a) and hif2α ATG-MO (b) embryos. (c and d) Lateral views of elavl3 expression in WT (c) and hif2α ATG-MO (d) 24 h.p.f. embryos. (e and f) Immunofluorescent staining of HuC/HuD (in red) in transverse brain sections of WT (e) and hif2α ATG-MO (f) 48 h.p.f. embryos. Nuclei are labeled by DAPI staining (in blue). (g and h) Transverse brain sections with TUNEL staining in birc5a ATG-MO (g) and birc5b ATG-MO (h) 48 h.p.f. embryos. Knockdown of birc5a and birc5b induced CNS apoptosis. (i and j) Lateral views of elavl3 expression in birc5a ATG-MO (i) and birc5b ATG-MO (j) 24 h.p.f. embryos. Compared with WT, birc5a and birc5b MO embryos exhibited lower elavl3 expression, and a similar pattern was found in hif2α MO embryos. (k and l) Immunofluorescent staining of HuC/HuD in transverse brain sections of birc5a ATG-MO (k) and birc5b ATG-MO (l) 48 h.p.f. embryos. The loss of neural differentiation in birc5a and birc5b MO embryos was confirmed by the deficiency in post-mitotic HuC/D expression. (m and n) Transverse brain sections with TUNEL staining in 48 h.p.f. hif2α ATG-MO embryos injected with 50 pg of birc5a mRNA (m) or birc5b mRNA (n). The intensive apoptosis observed in the hif2α morphants was effectively eliminated by either ectopic addition of birc5a or birc5b mRNA. (o and p), Lateral views of elavl3 expression in 24 h.p.f. hif2α ATG-MO embryos injected with 50 pg of birc5a mRNA (o) and birc5b mRNA (p). (q and r) Immunofluorescent staining of HuC/HuD in transverse brain sections of 48 h.p.f. hif2α ATG-MO embryos injected with 50 pg of birc5a mRNA (q) and birc5b mRNA (r). The loss of elavl3 expression in hif2α morphants was rescued by in vitro transcribed birc5a or birc5b mRNA

DNA content analysis by flow cytometry. For each sample, 30 000 events were acquired. In WT 48 h.p.f. embryos (a), 72, 25 and 3% of cells were in the G0/G1 (2n), S (2n∼4n) and G2/M (4n) phases, respectively; 5% apoptotic cells (<2n) were detected among the WT cells. hif2α (b) and birc5a (c) ATG-MO treatments increased the percentage of cells in S and G2/M compared with WT embryos. The percentage of apoptotic cells also increased by 3–4-fold compared with WT embryos. Treatment with 50 pg of ectopic birc5a mRNA (d) reduced hif2α ATG-MO-induced cell death to a level comparable to wild-type embryos. The percentage of cells in the S and G2/M phases also decreased significantly compared with hif2α and birc5a morphants

CNS defects in hif2α morphants are rescued by ectopic injection of survivin orthologue mRNA. The percentages of normal, moderate and severe (characterized by a smaller head size, short curved tail, narrow somites and massive CNS apoptosis) morphological phenotypes in the WT (n=50), hif1α ATG-MO (n=54), hif2α ATG-MO (n=69), hif2α SPL-MO (n=23), hif3α ATG-MO (n=25), hif1α, 3α dual ATG-MO (n=64), birc5a ATG-MO (n=45), birc5b ATG-MO (n=35) 48 h.p.f. embryos and hif2α ATG-MO co-injected with 50 pg birc5a mRNA (n=167) and in the hif2α ATG-MO embryos co-injected with 50 pg birc5b mRNA (n=262). Ectopic injection of either birc5a or birc5b mRNA restored most hif2α morphants to a normal phenotype whereas birc5a mRNA had better rescuing capability

Knockdown of birc5a and birc5b maintains neural progenitor cells in a proliferating stage and blocks maturation. (a–c) Lateral views of nes expression in WT (a), birc5a ATG-MO (b) and birc5b ATG-MO (c) 48 h.p.f. embryos. (d–f) Lateral views of pcna expression in WT (d), birc5a ATG-MO (e) and birc5b ATG-MO (f) 48 h.p.f. embryos. (g–i) Immunofluorescent staining of PCNA (in red) in transverse brain sections of WT (g), birc5a ATG-MO (h) and birc5b ATG-MO (i) 48 h.p.f. embryos. Nuclei are labeled by DAPI staining (blue). The high levels of nes and pcna expression following treatment with birc5a and birc5b MOs indicate that the NPCs are proliferating and not terminally differentiated. (j–o) Lateral views of cdkn1b (j–l) and cdkn1c (m–o) expression in WT (j and m), birc5a ATG-MO (k and n) and birc5b ATG-MO (l and o) 48 h.p.f. embryos. (p–r) Lateral views of ccnd1 expression in WT (p), birc5a ATG-MO (q) and birc5b ATG-MO (r) 48 h.p.f. embryos. The reduction in cdkn1b and cdkn1c expression and increase in ccnd1 expression in the CNS following treatment with birc5a and birc5b MOs suggest that the NPCs failed to exit from the cell cycle
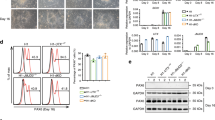
HIF2 α binds to birc5a and birc5b promoters. Since depletion of HIF2α resulted in a reduction in the transcription of both birc5a and birc5b, it raises a possibility that HIF2α indeed controls the survivin orthologues through direct interactions with their promoters. We retrieved the upstream sequence of both bric5a and birc5b genes from UCSC genome website (http://genome.ucsc.edu/) and identified multiple hypoxia-response element (HRE) core sequences (A/GCGTG) in the promoter regions (Figure 10c). Similar multiple HRE core sequences were also identified in human and mouse BIRC5 promoters (Figure 10c). To determine if zebrafish HIF2α binds to the HRE specifically, oligonucleotides containing the HRE core sequence ACGTG were synthesized, biotin labeled and electrophoretic mobility shift assay (EMSA) was carried out (Figure 10a). When HIF2α was expressed alone or it was coexpressed with ARNT1a in COS-1 cells, a mobility complex was formed with wild-type HRE probe (lanes 3 and 4). This mobility complex was effectively competed by a 200-fold molar excess of the unlabeled HRE (lane 5). Conversely, the mobility complex was, however, unaffected by a 200-fold molar excess of the unlabeled mutated HRE (lane 6). These observations demonstrated the transfected HIF2α binds to HRE with a sequence specificity.

HIF2α binds to birc5a and birc5b promoter. (a) HIF2α binds to HRE as confirmed by EMSA. COS-1 cells were transfected with zHif2A-HA and zARNT1A-HA as indicated. Nuclear extracts were collected at 48 h post-transfection followed by gel mobility shift assays by using the HRE oligonucleotides as a probe. The asterisks in lanes 3, 4 and 6 indicate the specific HIF2–HRE complex formed. NE, nuclear extract; COS-1, nuclear extract of COS-1 cells without transfection as a control; Hif2A, nuclear extract of COS-1 cells transfected with pcDNA3-zHif2A-HA; Hif2A+zARNT1A, nuclear extract of COS-1 cells co-transfected with pcDNA3-zHif2A-HA and pcDNA3-zARNT1A-HA. (b) HIF2α binds to the birc5a and birc5b promoters in brain tissues under physiological conditions. A chromatin immunoprecipitation (ChIP) assay was performed with polyclonal antibody against anti-HIF2α or preimmune serum as control. Chromatin was prepared from adult brain tissue and then immunoprecipitated using polyclonal antibody against anti-HIF2α or preimmune serum (IgG) as control. The immunoprecipitates were subjected to PCR using specific primers spanning the HREs in the birc5a and birc5b promoters (shown in panel c). The module 5 of birc5a and modules 2 and 6 of birc5b were significantly enriched by the HIF2α antibody. The rest modules failed to be amplified. A portion of chromatin solution (2.4%) was used as input. The primers used in ChIP assay were listed in Table 1. (c) Schematic representation of the birc5a (NM_194397), birc5b (NM_145195), human BIRC5 (NM_001012271) and mouse Birc5 (NM_001012273) promoters. The putative binding sites for HIF2, including HRE1 (ACGTG, black bars) and HRE2 (GCGTG, gray bars), are indicated. The position of the DNA modules analyzed in ChIP-PCR assays is indicated by the boxes. The amplified fragments in ChIP assays (panel b), including the module 5 in birc5a promoter and the modules 2 and 6 in birc5b promoter, are indicated by black boxes
To examine whether HIF2α binds directly to birc5a and bric5b promoter in vivo, we immunoprecipitated crosslinked chromatin from adult brain tissue with a HIF2α-specific antibody. The precipitated chromatin was then analyzed by PCR using primer pairs that amplified modules spanning the birc5a and bric5b loci. The ChIP assay demonstrated that the module 5 of birc5a and modules 2 and 6 of birc5b were significantly enriched by the HIF2α antibody (Figures 10b and c), suggesting that HIF2α binds directly to both birc5a and bric5b promoters to modulate their expressions.
Discussion
HIFα and survivin are prominent markers in many human cancers and have critical roles in cell apoptosis and proliferation. Previously, we found that depletion of all of the HIF α factors caused massive apoptosis in the brains of zebrafish embryos.20 Here, we showed that of the three HIF-α factors, HIF2α is the factor that modulates neural progenitor cell growth and differentiation through its downstream effects on the survivin orthologues, Birc5a and Birc5b. Depletion of HIF2α expression caused reductions in both of the birc5a and birc5b transcripts, which in turn elicited massive apoptosis and abrogated neural progenitor cell differentiation. Although human survivin (BIRC5) and zebrafish survivin orthologues (birc5a and birc5b) promoters are all enriched with multiple HREs, these survivin genes are modulated by different HIF α factors. In human cancer cells, BIRC5 transcription is modulated by HIF1α through its direct binding to the HRE in the proximate promoter region.23 In zebrafish, we demonstrate that both birc5a and birc5b transcriptions are controlled by HIF2α, but not HIF1α. It is noteworthy that although zebrafish HIF1α does not involve in NPC growth and differentiation, it still have an essential role in other developmental events. Depletion of HIF1α expression abrogated erythropoiesis and reduced hemoglobin content (data not shown). It suggests that the sets of HIF1α- and HIF2α-modulated genes in human and zebrafish are slightly different due to the discriminative target selectivity of HIF1α and HIF2α factors cross species.
It has been shown that HIFs have important roles in embryonic cell differentiation and proliferation.24 Hypoxic preconditioning promotes embryonic stem cell neural differentiation and enhances cell survival through upregulating HIF1α and HIF2α.25 However, the functions of these HIFs in cell differentiation still have not been elucidated. Here, we demonstrate that HIF2α depletion first causes massive NPC apoptosis and then stimulates the remaining NPCs proliferation. Nevertheless, these newly produced NPCs did not undergo final differentiation. Ectopic treatment with the birc5a or birc5b mRNA not only successfully prevented hif2α MO-induced apoptosis and morphological abnormality, but also restored neural differentiation. It suggests that the abrogation of differentiation is a secondary effect elicited by HIF2α depletion due to the loss of its downstream effectors, Birc5a and Birc5b.
Previous studies have demonstrated a close relationship between the hypoxia-signaling pathway and cell-cycle arrest in mammalian cells. For instance, either overexpression of the stable forms of HIF1α and HIF2α in murine cells or treatment of murine cells with hypoxia directly resulted in an increase in p27 transcription and cell-cycle arrest at the G1 phase.26, 27 Moreover, targeted depletion of HIF1α in chondrocytes resulted in decreased p57 expression, enhanced BrdU incorporation and cell death.28 We demonstrate that the loss of survivin could be responsible for the reductions in p27 and p57 expression and the aberrant neural development that is observed in the hif2α morphant zebrafish embryos. Survivin also had a similar effect on cell differentiation in mouse erythroid development. Depletion of survivin from hematopoietic progenitor cells results in aberrant erythroid formation.29 Likewise, earlier studies have demonstrated that depletion of zebrafish birc5a and birc5b transcription evoked profound neuro-developmental, hematopoietic, cardiogenic, vasculogenic and angiogenic defects.22
In Drosophila, dying cells trigger compensatory proliferation through both initiator and effector caspase activities.30 In apoptotic proliferating tissues, the initiator caspase Dronc coordinates apoptosis and compensatory proliferation through the Jun N-terminal kinase and p53 pathways, which in turn induce secretion of the mitogens Decapentaplegic (Dpp) and Wingless (Wg).30, 31 In apoptotic differentiating tissues, the effector caspases DrICE and Dcp-1 activate the hedgehog pathway to induce compensatory proliferation.32 Hedgehog signaling triggers the reentry of cells that had previously exited the cell cycle.32, 33 In zebrafish embryos, knockdown of hif2α resulted in the loss of expression of the survivin orthologues, which in turn caused severe neural progenitor cell death, increased proliferation activity and abrogated neural differentiation. This phenomenon of cell death-associated proliferation and abrogation of differentiation in zebrafish embryos is analogous to what has been observed in Drosophila apoptosis-induced compensatory proliferation. We hypothesize that reentry into the cell cycle causes descendant cells to remain in the progenitor stage without further differentiating. These findings suggest that a key function of HIF2α in developing embryos is related to inhibition of neural progenitor cell death and maintenance of neural differentiation through the functions of downstream survivin orthologues (Figure 11). Depletion of HIF2α and the survivin orthologues causes massive neural progenitor cell death, which induces compensatory proliferation and abrogates neural differentiation.

A proposed mechanism by which HIF2α deprivation induces NPC apoptosis and blocks neural differentiation. Knockdown of hif2α results in a reduction in the survivin orthologues, which in turn causes apoptosis and induces complementary proliferation in neighboring cells. The replenished proliferating cells remain in the cell cycle and do not terminally differentiate
It has been demonstrated that inhibition of human HIF2α promotes tumor cell death through p53 activation. HIF2α depletion promotes p53-mediated responses in clear cell renal cell carcinoma (ccRCC) by disrupting cellular redox homeostasis, which thereby permits ROS accumulation and DNA damage.14 Although a similar level of p53 accumulation was also observed in hif2α morphant embryos (data not shown), we demonstrated that inhibition of p53 expression did not prevent massive apoptosis in hif2α morphant embryos. Similarly, HIF2α inhibition-induced massive apoptosis was also observed in the p53 mutant line. Instead, we demonstrated that the massive apoptosis observed in hif2α morphant embryos is caused by depletion of the survivin orthologues. Ectopic treatment with survivin mRNAs rescued the defects induced by HIF2α inhibition, indicating that survivins act downstream of HIF2α to maintain cell survival. It is noteworthy that expression of survivin/BIRC5 in ccRCC was inversely correlated with cancer-specific survival.34 It will be interesting to examine the correlation between HIF2α and survivin expression in ccRCC patients. Our results have shed light on how cancer cells protect themselves through the HIF and survivin pathways.
Materials and Methods
Morpholino and mRNA injection
Adult zebrafish were maintained and staged as described previously.35 The tp53M214K fish line generated by the laboratory of Thomas Look36 was obtained from the Taiwan Zebrafish Core Facility in Taiwan. All embryos were grown in 0.003% 1-phenyl-2-thiourea after 14 h.p.f. at 28.5°C to block pigmentation and mediate visualization. Morpholinos (Gene Tools, Philomath, OR, USA) were designed against Danio rerio hif1α, hif2α and hif3α (GenBank accession AY326951, DQ375242 and AY330295, respectively). The birc5a, birc5b and p53 ATG-MOs were designed as described previously.30, 37 The translation-blocking morpholino (ATG-MO) sequences and dosages applied were as follows: hif1α ATG-MO, 5′-CAGTGACAACTCCAGTATCCATTCC (6 ng); hif2α ATG-MO, 5′-CGCTGTTCTCGCGTAATTCCCGCAG-3′ (6 ng); hif3α ATG-MO, CCTTTTCGACGTAGAGTTCACCATC (12 ng); birc5a ATG-MO, 5′-TGCAAGATCCATTTTGTGGGAGGTT-3′ (4 ng); birc5b ATG-MO, 5′-GAAGTCTTTTTTCATAACTATACAT-3′ (12 ng); p53 ATG-MO 5′-GCGCCATTGCTTTGCAAGAATTG-3′ (9 ng). To confirm the specificity of the hif2α ATG-MO, a second morpholino that blocks pre-mRNA splicing of hif2α at the exon 2–intron 2 junction was also designed: hif2α SPL-MO, 5′-GACTGTGTACCTGAGTTGACGAGTT-3′ (6 ng). Aliquots of the morpholinos (4.6 nl each) were injected into 1–2 cell-stage embryos using a Drummond Nanoject (Drummond, Broomall, PA, USA). For capped RNA synthesis, the full-length cDNAs of hif2α, birc5a and birc5b were cloned into the pT7TS vector. After linearization, the plasmids were transcribed in vitro with T7 RNA polymerase and the mMESSAGE mMACHINE kit from Ambion (Austin, TX, USA). For the rescue assay, 75 pg of purified hif2α mRNA or 50 pg of purified birc5a and birc5b mRNAs were co-injected with hif2α ATG-MO into 1–2 cell-stage embryos.
In situ hybridization
The hif2α (DQ375242, bases 2557–3137), birc5a (NM_194397, bases 38–485), birc5b (NM_145195, bases 2–421), elavl3/HuC (BC065343, bases 55–554), nes (XM_001919887, bases 54–754), pcna (BC064299, bases 255–1367), p27/cdkn1b (NM_212792, bases 611–1262), p57/cdkn1c (NM_001002040, bases 50–1133) and ccnd1 (NM_131025, bases 1094–2213) templates were amplified from cDNA and cloned into the pGEM-T-easy vector (Promega, Madison, WI, USA). To generate probes, the linearized plasmids were used as templates for in vitro transcription with the DIG RNA labeling kit (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer's instructions. Whole-mount in situ hybridization was performed essentially as described previously.35 Nitro blue tetrazolium/bromo-4-chloro-3-indolyl phosphate (Roche Applied Science) served as the substrate for color development. Double fluorescent in situ hybridization38 was performed using DIG-UTP and FITC-UTP-labeled RNA probes followed by sequential detection using HRP-labeled anti-DIG and HRP-labeled anti-FITC antibodies (Roche, Indianapolis, IN, USA) and fluorescent tyramide amplification. Following RNA probe hybridization, embryos were blocked in 10% serum, 1% BSA and were then incubated with HRP anti-FITC at 1 : 500 dilution for overnight in 10% serum, 1% BSA. After washing, anti-FITC antibody was visualized with TSA Plus Fluorescein Solution (Perkin-Elmer, Waltham, MA, USA) for 45 min, dehydrated in methanol and quenched with 1% H2O2 in methanol for 30 min. After rehydrating, embryos were blocked again and incubated with HRP anti-DIG at 1 : 500 dilution for 4 h, then washed and reacted with TSA Plus Cy3 solution (Perkin-Elmer) for 45 min. Embryos were cleared in glycerol, mounted and viewed on a Zeiss LSM 510 confocal fluorescence microscope (Zeiss, Jena, Germany).
Flow cytometric DNA content analysis
After removal of the chorion and yolk, 20 embryos were washed in PBS and transferred to a collection tube. Embryos were digested with a trypsin (0.05%)/EDTA (1 mM) solution for 10 min at room temperature and then completely dissociated into single-cell suspensions by pipetting. Trypsin was inactivated by incubation with CaCl2 (1 mM)/lamb serum (5%), and the whole suspension was passed through a 40-μm filter (BD Falcon, Franklin Lakes, NJ, USA). The cells were centrifuged at 2000 r.p.m. at 4°C, washed in PBS and fixed in 70% ethanol overnight at −20°C. After fixation, the cells were centrifuged and resuspended in PBS. The suspended cells were stained by treatment with RNase A (200 μg/ml) and propidium iodide (50 ng/ml) (Sigma, St. Louis, MO, USA) for at least 15 min at 4°C in the dark. In all, 30 000 events were acquired per sample and propidium iodide-positive cells were quantified by flow cytometry (BD FACSCanto, Franklin Lakes, NJ, USA). Cells with DNA content less than that of 2 N (G0/G1) cells were classified as apoptotic cells and cells distribute between 2 and 4 N (G2/M) cells were S-phase cells.
RT-qPCR
Total RNA extracted from embryos using TRIzol (Invitrogen, Carlsbad, CA, USA) followed by DNase I treatment (Roche) was used for cDNA synthesis with Superscript-II (Invitrogen). Quantitative (q)PCR was performed using the Power SIBER Q-PCR Master Mix (BioNoVas, Toronto, Canada) in a Bio-Rad, iQ5 Gradient Real Time PCR System (Bio-Rad, Hercules, CA, USA). qPCR conditions were 95°C for 3 min and then 40 cycles of 95°C for 15 s and 60°C for 1 min. The following primers were designed using the Primer Express Software version 3.0 (Applied Biosystems, Carlsbad, CA, USA):
birc5a forward, 5′-GTGGCCACGCGATTGAA-3′;
birc5a reverse, 5′-CAGTCTGGATGATCTGGTTTCTCTT-3′;
birc5b forward, 5′-GGAGCGACTTCGCATCTACAT-3′;
birc5b reverse, 5′-ACCTCATCACGAAAGTAGGCAATC-3′;
cdkn1b forward, 5′-TGGGTCGTGCCACCAATTA-3′;
cdkn1b reverse, 5′-TGCATCACGTTTCACCACAGA-3′;
cdkn1c forward, 5′-AACAGCACGCCGCAAATT-3′;
cdkn1c reverse, 5′-TCGACTCCACGACCCTCTTT-3′;
ccnd1 forward, 5′-TGTGATGAATCTCTTCGTCTTTGTT-3′;
ccnd1 reverse, 5′-GTCCCATGACGCCTTTGG-3′;
β-actin forward, 5′-CCCCGAGAGGACAACAATGTA-3′;
β-actin reverse, 5′-TGAGGAGGGCAAAGTGGTAAA-3′.
Duplicate mean values were calculated according to the Ct quantification method using β-actin transcript levels as the reference for normalization. Relative quantification was determined using the ÄÄCt method: relative expression=2−ΔΔCt.
Apoptosis detection
Zebrafish embryos at 24 h.p.f. were dechorionated and incubated for 30 min in 5 μg/ml acridine orange in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2 and 0.33 mM MgSO4). The embryos were subsequently washed repeatedly in E3 medium. For microscopic examination, embryos were transferred into 0.01% ethyl 3-aminobenzoate methanesulfonate (tricaine, Sigma). TUNEL assays were performed using an in situ cell death detection kit (POD, Roche) as described by the manufacturer. For combined fluorescent nes DIG in situ/TUNEL staining, embryos were first in situ hybridized with fluorescence nes RNA probe followed by TUNEL assay.39 The protocol of fluorescence in situ hybridization is as described above. The RNA probe was labeled by DIG. After conjugated with HRP anti-DIG antibody (Perkin-Elmer), the signal was detected by TSA Plus Fluorescein Solution (Perkin-Elmer). This was followed by TUNEL assay. The embryos were manually de-yolked and the images were captured on a Zeiss LSM 510 confocal microscope.
Chromatin immunoprecipitation
ChIP was performed by using Magna ChIP Protein A magnetic beads (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, adult brain tissues were crosslinked with formaldehyde and chromatin was isolated. The isolated chromatin was sonicated to an average size of about 200–300 bp. Protein A magnetic beads were incubated with antibody against HIF2α (ChIP grade rabbit polyclonal antibody purchased from GeneTex (Irvine, CA, USA), GTX103707) or control IgG at 4°C for 1 h. Immunoprecipitation reactions were performed by incubating magnetic beads-linked antibody with the chromatin overnight at 4°C. The immunoprecipitated chromatin complexes were washed with TE buffer and the protein–DNA crosslinks were reverse crosslinked by proteinase K digestion at 62°C for 2 h followed by 95°C for 10 min. The DNA was purified by spin columns and then eluted with elution buffer. Immunoprecipitated DNA and input DNA were used as templates for PCR amplification (30 cycles). The PCR primers used in ChIP assay are listed in Table 1.
Electrophoretic mobility shift assay
After COS-1 cells were transfected with the indicated plasmids for 48 h, nuclear extracts were prepared by minipreparation method as described previously.40 The nuclear extracts were preserved at −70°C until use. Oligonucleotide probe containing the HRE core sequence (5′-CATCGTCCTTCAAACGTGTAACTAAGTTCA-3′) and mutant probe (5′-CATCGTCCTTCAAGTACATAACTAAGTTCA-3′) was synthesized and labeled with 5′-end biotin (Purigo, Taipei, Taiwan). For each EMSA reaction, 20 μg of nuclear extracts from normal or transfected COS-1 cells was mixed with biotin-labeled probes and premixed binding reagents (Thermo, Waltham, MA, USA) and incubated at room temperature for 30 min. For competitive experiments, 50 × normal and mutant probes were added. Unbound DNA probes were resolved from protein–DNA complexes by electrophoresis on a 10% polyacrylamide gel. After electrophoresis, DNA oligonucleotides were transferred onto a nylon membrane (Pall, Washington, NY, USA) and UV crosslinked. Biotin-labeled DNAs were detected by using the Lightshift Chemiluminescent EMSA Kit according to the manufacturer's instructions (Thermo).
Immunostaining
Zebrafish acetylated tubulin IHC was performed as described in The Zebrafish Book.35 Briefly, embryos were fixed in 4% paraformaldehyde (PFA) at 4°C, dehydrated and stored in MeOH. After rehydration, embryos were treated with proteinase K, fixed in 4% PFA, washed in 0.3% Triton X-100 in PBS (PBT) and subsequently blocked for 1 h in PBT containing 5% horse serum. Embryos were incubated with mouse anti-acetylated tubulin antibody (1 : 1000, Sigma T-6793).30 After several washes with PBT, embryos were blocked for 1 h and incubated with goat anti-rabbit Alexa Fluor 568 secondary antibody (Sigma). Images were acquired using a Zeiss Axioskop 2 fluorescence microscope (Zeiss). For cross-sectional analysis,35 embryos were embedded in OCT mount medium (Sakura Finetek, Torrance, CA, USA), Cryostat sectioned (CM-1900, Leica, Nussloch, Germany) and mounted on glass slides (Superfrost Plus, J1800AMNZ). The primary antibodies included mouse anti-HuC/HuD (Molecular Probes, Eugene, OR, USA, 1 : 1000) and monoclonal anti-PCNA (Sigma, P8825, 1 : 1000). After several washes with PBT, the slides were incubated with goat anti-rabbit Alexa Fluor 568 secondary antibody (Sigma). After repeated washes, sections were counterstained with 4,6-diamidino-2-phenylindole (DAPI, Sigma) at 10 ng/ml to visualize the nuclei. Fluorescence and UV light images were acquired using a fluorescence microscope (OLYMPUS BX 51, Olympus, Tokyo, Japan).
Accession codes
Abbreviations
- AO stain:
-
acridine orange stain
- AOE:
-
anti-oxidant enzyme
- ascl1b:
-
achaete-scute complex-like 1b
- BIR domain:
-
baculovirus IAP repeat domain
- ccnd1:
-
cyclin D1
- ccRCC:
-
clear cell renal cell carcinoma
- cdkn1b/1c:
-
cyclin-dependent kinase inhibitor 1b (p27/kip1)/1c (p57/kip2)
- ChIP:
-
chromatin immunoprecipitation
- CNS:
-
central nervous system
- DAPI:
-
4′,6-diamidino-2-phenylindole
- Dpp:
-
decapentaplegic
- elavl3:
-
ELAV (embryonic lethal, abnormal vision, Drosophila)-like 3 (Hu antigen C)
- EMSA:
-
electrophoretic mobility shift assay
- HIF:
-
hypoxia-inducible factor
- HuC/D:
-
Hu antigen C/D
- IAP:
-
inhibitor of apoptosis protein
- MO:
-
morpholino oligonucleotide
- NPCs:
-
neuronal progenitor cells
- PCNA:
-
proliferation cell nuclear antigen
- ROS:
-
reactive oxygen species
- VHL:
-
von Hippel–Lindau protein
- TUNEL:
-
terminal transferase dUTP Nick End Labeling
- Wg:
-
wingless
References
Lopez-Barneo J, Pardal R, Ortega-Saenz P . Cellular mechanisms of oxygen sensing. Annu Rev Physiol 2001; 63: 259–287.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399: 271–275.
Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N . Expression and characterization of hypoxia-inducible factor (HIF)-3 alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3 alpha. Biochem Biophys Res Commun 2001; 287: 808–813.
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH et al. Cellular and developmental control of O-2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 1998; 12: 149–162.
Ryan HE, Lo J, Johnson RS . HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 1998; 17: 3005–3015.
Yoon D, Pastore YD, Divoky V, Liu EL, Mlodnicka AE, Rainey K et al. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem 2006; 281: 25703–25711.
Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H et al. Loss of HIF-2 and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 2002; 8: 702–710.
Peng J, Zhang LY, Drysdale L, Fong GH . The transcription factor EPAS-1/hypoxia-inducible factor 2 alpha plays an important role in vascular remodeling. Proc Natl Acad Sci USA 2000; 97: 8386–8391.
Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1(−/−) mice. Nat Genet 2003; 35: 331–340.
Tomita S, Ueno M, Sakamoto M, Kitahama Y, Ueki M, Maekawa N et al. Defective brain development in mice lacking the Hif-1 alpha gene in neural cells. Mol Cell Biol 2003; 23: 6739–6749.
An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM . Stabilization of wild-type p53 by hypoxia-inducible factor 1 alpha. Nature 1998; 392: 405–408.
Bruick RK . Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA 2000; 97: 9082–9087.
Dong Z, Venkatachalam MA, Wang JZ, Patel Y, Saikumar P, Semenza GL et al. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia – HIF-1-independent mechanisms. J Biol Chem 2001; 276: 18702–18709.
Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B et al. HIF2 alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci USA 2009; 106: 14391–14396.
Ambrosini G, Adida C, Altieri DC . A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997; 3: 917–921.
Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL et al. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol 2000; 10: 1319–1328.
Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC . Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol 1998; 152: 43–49.
Jiang YY, de Bruin A, Caldas H, Fangusaro J, Hayes J, Conway EM et al. Essential role for survivin in early brain development. J Neurosci 2005; 25: 6962–6970.
Altieri DC . Validating survivin as a cancer therapeutic target. Nat Rev Cancer 2003; 3: 46–54.
Hsieh CS, Ko CY, Chen SY, Liu TM, Wu JS, Hu CH et al. In vivo long-term continuous observation of gene expression in zebrafish embryo nerve systems by using harmonic generation microscopy and morphant technology. J Biomed Opt 2008; 13: 064041.
Rojas DA, Perez-Munizaga DA, Centanin L, Antonelli M, Wappner P, Allende ML et al. Cloning of hif-1 alpha and hif-2 alpha and mRNA expression pattern during development in zebrafish. Gene Expr Patterns 2007; 7: 339–345.
Delvaeye M, De Vriese A, Zwerts F, Betz I, Moons M, Autiero M et al. Role of the 2 zebrafish survivin genes in vasculo-angiogenesis, neurogenesis, cardiogenesis and hematopoiesis. BMC Dev Biol 2009; 9 ARTN 25.
Peng XH, Karna P, Cao ZH, Jiang BH, Zhou MX, Yang L . Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1 alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression. J Biol Chem 2006; 281: 25903–25914.
Mazumdar J, Dondeti V, Simon MC . Hypoxia-inducible factors in stem cells and cancer. J Cell Mol Med 2009; 13: 4319–4328.
Francis KR, Wei L . Human embryonic stem cell neural differentiation and enhanced cell survival promoted by hypoxic preconditioning. Cell Death Dis 2010; 1: e22.
Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS . Hypoxia-inducible factor 1 alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 2003; 23: 359–369.
Hackenbeck T, Knaup KX, Schietke R, Schodel J, Willam C, Wu XQ et al. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle 2009; 8: 1386–1395.
Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS . Hypoxia in cartilage: HIF-1 alpha is essential for chondrocyte growth arrest and survival. Genes Dev 2001; 15: 2865–2876.
Gurbuxani S, Xu YF, Keerthivasan G, Wickrema A, Crispino JD . Differential requirements for survivin in hematopoietic cell development. Proc Natl Acad Sci USA 2005; 102: 11480–11485.
Ryoo HD, Gorenc T, Steller H . Apoptotic cells can induce compensatory cell proliferation through the JNK and the wingless signaling pathways. Dev Cell 2004; 7: 491–501.
Wells BS, Yoshida E, Johnston LA . Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol 2006; 16: 1606–1615.
Fan Y, Bergmann A . Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell 2008; 14: 399–410.
Fan Y, Bergmann A . Apoptosis-induced compensatory proliferation. The cell is dead. Long live the cell!. Trends Cell Biol 2008; 18: 467–473.
Kosari F, Parker AS, Kube DM, Lohse CM, Leibovich BC, Blute ML et al. Clear cell renal cell carcinoma: gene expression analyses identify a potential signature for tumor aggressiveness. Clin Cancer Res 2005; 11: 5128–5139.
Westerfield M (ed.). The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). 4th edn. University of Oregon Press: Eugene, 2000.
Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CDM et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci USA 2005; 102: 407–412.
Ma ACH, Lin R, Chan PK, Leung JCK, Chan LYY, Meng A et al. The role of survivin in angiogenesis during zebrafish embryonic development. BMC Dev Biol 2007; 7 ARTN 50.
Brend T, Holley SA . Zebrafish whole mount high-resolution double fluorescent in situ hybridization. J Vis Exp 2009: (25); http://www.jove.com/index/Details.stp?ID=1229, doi:10.3791/1229.
Chen HL, Yuh CH, Wu KK . Nestin is essential for zebrafish brain and eye development through control of progenitor cell apoptosis. PLoS One 2010; 5: e9318.
Deryckere F, Gannon F . A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques 1994; 16: 405.
Acknowledgements
We thank SY Chen and CT Shih (NTOU) for critical discussions about the manuscript, J Chang (NTOU) for statistical analysis, KY Huang, WH Liao (Academia Sinica) for confocal microscope photograph, MS Yu (Taiwan Zebrafish Core Facility, NSC 99-2321-B-400-0001) for providing the tp53M214K fish and YC Cheng (CGU) for reagents and fish lines. This study was supported by grants from the National Science Council (Taiwan), the Ministry of Education and the Center for the Excellence of Marine Bioenvironmental and Biotechnology (NTOU) to CHH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by N Chandel
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ko, CY., Tsai, MY., Tseng, WF. et al. Integration of CNS survival and differentiation by HIF2α. Cell Death Differ 18, 1757–1770 (2011). https://doi.org/10.1038/cdd.2011.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.44
Keywords
This article is cited by
-
(H)IF applicable: promotion of neurogenesis by induced HIF-2 signalling after ischaemia
Pflügers Archiv - European Journal of Physiology (2021)
-
Hypoxia-inducible factor-2α is crucial for proper brain development
Scientific Reports (2020)
-
Functional enrichment analysis based on long noncoding RNA associations
BMC Systems Biology (2018)
-
Zebrafish cyclin Dx is required for development of motor neuron progenitors and its expression is regulated by hypoxia-inducible factor 2α
Scientific Reports (2016)
-
Genome-wide mapping of Hif-1α binding sites in zebrafish
BMC Genomics (2015)
