
Abstract
Despite an abundance of literature on the role of caspase-2 in apoptosis, there exists much controversy about this protease making it difficult to place caspase-2 correctly in the apoptotic cascade, and hence its role in apoptosis remains unclear. The identification of the PIDDosome as a signaling platform for caspase-2 activation prompted intense investigation into the true role of this orphan caspase. What has emerged is the idea that caspase-2 may not be mandatory for apoptosis and that activation of this caspase in response to some forms of stress has other effects on the cell such as regulation of cell cycle progression. This idea is particularly relevent to the discovery that caspase-2 may act as a tumor suppressor. Here, we discuss the proposed mechanisms through which caspase-2 signals, in particular those involving PIDD, and their impact on cellular fate.
Similar content being viewed by others


Facts
-
Caspase-2 is an initiator caspase because it is activated by dimerization.
-
The PIDDosome is a signaling platform for caspase-2 activation that contains the proteins PIDD (p53-induced protein with a death domain) and RAIDD (RIP-associated ICH-1/CAD-3 homologous protein with a death domain).
-
PIDD induces caspase-2 activation and nuclear factor κB (NFκB) activation in response to DNA damage.
-
Caspase-2 can induce apoptosis and cell cycle arrest and may function as a tumor suppressor.
-
Caspase-2-deficient mice have features consistent with accelerated aging.
Open Questions
-
Is the primary function of caspase-2 apoptotic?
-
Which proteolytic substrates of caspase-2 are important for its role in cell cycle arrest?
-
Do alternative caspase-2 activating platforms exist that are independent of PIDD and/or RAIDD?
Caspase-2 is a member of the cysteine aspartate protease family known as caspases. Caspases are generally responsible for the initiation and execution of apoptosis although a subset of caspases are primarily involved in the regulation of inflammation.1 When caspases are activated in cells, they cleave a wide variety of specific protein substrates resulting in the classical morphological characteristics of apoptosis, including DNA fragmentation, chromatin condensation and plasma membrane blebbing. Caspase-2 is the most evolutionarily conserved of all caspases2 and although there is a wealth of literature on the role of caspase-2 in apoptosis, there is much controversy regarding this protein, making it difficult to correctly place caspase-2 in the apoptotic cascade and hence its role in apoptosis remains unclear, making it the orphan of the caspase family.3, 4
Of the many important contributions Jürg Tschopp made to the apoptosis field, his work on identifying and characterizing the caspase-2 signaling platform has been vital to advancing our understanding of the function of this poorly defined caspase and dispelling some of the controversies that have existed. This review highlights these important discoveries Jürg Tschopp made regarding caspase-2 activation and function in the context of a discussion of the more recent advancements in the caspase-2 field.
Caspase-2 is Activated by Dimerization
Caspases can be divided into two distinct groups, the initiator caspases, including caspase-8 and -9, and the executioner caspases, including caspase-3 and -7.5 The initiator caspases generally function at the apex of their respective signaling cascades and promote activation of executioner caspases either directly or indirectly. Initiator caspases are present in the cell as inactive monomers and their activation is promoted by dimerization. Dimerization results when initiator caspases are recruited to large molecular weight protein complexes that act as signaling platforms.6 For example, genotoxic stress engages the mitochondrial pathway resulting in cytochrome c release from mitochondria, which in turn activates the apoptotic protease-activating factor-1 (APAF-1) apoptosome complex that recruits and activates caspase-9. Following activation, caspase-9 directly activates the executioner caspase, caspase-3.7
Although caspase-2 shares some substrate specificity with the executioner caspases,8, 9 in many ways it is considered to be an initiator caspase. All caspases are synthesized as single chain pro-caspases, which undergo intra-chain cleavage to generate large and small catalytic subunits. However, although cleavage is necessary and sufficient to activate executioner caspases, evidence shows that dimerization is required for the activation of initiator caspases and the subsequent intra-chain cleavage serves to stabilize the active enzyme.5
Caspase-2 is activated in a similar way. Caspase-2 has no detectable activity in its monomeric form regardless of its cleavage status, while a dimeric form of a cleavage defective mutant of caspase-2 retains up to 20% of the enzyme's activity. Once the dimerized caspase undergoes autoprocessing, the enzyme becomes fully active.10 Thus dimerization is the initiating step in caspase-2 activation.
Recruitment of caspase-2 to a higher molecular weight protein complex has been demonstrated in cell extracts.11 This occurred in a similar fashion to caspase-9 but, importantly, this complex was distinct from the APAF-1 apoptosome and was activated by temperature shift (cell extracts were heated to 37 °C for 30 min) rather than by cytochrome c. This was the first indication that a specific signaling complex for caspase-2 exists and further evidence that caspase-2 behaves like an initiator caspase.
Identifying the Caspase-2 Signaling Platform
Caspase-2 has a similar domain organization to the initiator caspases, caspase-8 and -9. It contains a long prodomain, and a large and small catalytic subunit12 (Figure 1). The prodomain of caspase-2 contains a protein:protein interaction motif termed a caspase recruitment domain (CARD).13 The term CARD was first coined by Jürg Tschopp when he identified a conserved protein:protein interaction domain present in several caspases and caspase adaptor proteins, namely caspase-2, RAIDD and the Caenorhabditis elegans protein CED3.13 Upon proposing this domain he postulated:
‘The biological role of proteins containing this domain allows the prediction of a network of interactions necessary to recruit caspases to the receptor complexes signaling apoptosis.13’

Domain organization of the PIDDosome components. The PIDDosome is comprised of the proteins PIDD, RAIDD and caspase-2. The cleavage fragments resulting from PIDD autoprocessing are indicated
Since then, a large array of proteins have been identified as CARD-containing proteins and although not all of them bind caspases, the central idea that CARDs mediate recruitment to signaling complexes holds true.14 This is clearly exemplified in the caspase-2 pathway. The CARD in caspase-2 binds to the CARD present in RAIDD and thus RAIDD acts as an adaptor protein, recruiting caspase-2 to the signaling complex.15, 16
When RAIDD was identified, it was initially proposed to be a component of the tumor necrosis factor receptor 1 (TNFR1) signaling complex.15 It was shown to bind to receptor interacting protein kinase 1 (RIPK1) and it was thought that caspase-2 could be recruited to the TNFR1–TRADD–RIPK1 complex in response to tumor necrosis factor (TNF) stimulation. However, there is little evidence that caspase-2 is required for TNF-induced apoptosis and RAIDD-deficient cells do not show any resistance to TNF indicating that an alternative signaling complex for caspase-2 exists.17
It was Tschopp's laboratory that discovered an important missing piece of this puzzle. They identified PIDD as an essential component of the caspase-2 activation complex that was assembled upon temperature shift of cell extracts and hence was termed the PIDDosome.18
The PIDDosome
PIDD, also known as LRDD (leucine-rich repeat and death domain-containing protein), was originally identified as a p53 inducible gene.19, 20 PIDD contains a death domain (DD) at its carboxyl terminal, two ZU5 interaction domains and seven tandem leucine-rich repeats (LRRs) at the amino terminal18 (Figure 1). The DD is a protein:protein interaction domain that is similar to the CARD.21 ZU5 domains are found in the ZO-1, ankyrin and Unc5-like netrin receptors but their function is unclear.22, 23 LRRs represent another class of protein interaction motif that is found in a variety of proteins with diverse functions.24 LRRs exist in a number of nucleotide-binding oligomerization domain (NOD)-like and NALP-like proteins (for reviews on Jürg Tschopp's contributions to this interesting area of research see elsewhere in this issue), and it is this motif that recognizes various pathogen-associated molecular patterns and danger-associated molecular patterns that induce inflammasome or NODosome assembly resulting in caspase-1 activation or NFκB activation, respectively.25, 26 PIDDosome assembly is likely triggered in an analogous manner and when Tschopp presented the first description of the PIDDosome he hypothesized as such:
‘The ultimate trigger leading to assembly seems to be a stress-related signal of unknown nature.18’
Unfortunately, we have yet to prove this hypothesis because, at present, a molecule that can bind the LRRs in PIDD has not been identified. However, consistent with this idea, Tschopp and colleagues27 also demonstrated that cells expressing a PIDD mutant lacking the N-terminal LRRs showed enhanced function. This suggests that the LRRs act as an autoinhibitory function similar to the WD repeats in APAF-128 or the LRRs in NALP or NOD proteins.29
Assembly of the PIDDosome complex occurs when PIDD binds RAIDD via a DD present in both proteins. The core of the complex consisting of the DD of PIDD and the DD of RAIDD has been crystallized, and this structure revealed that the complex is comprised of five PIDD DDs and seven RAIDD DDs, which form two stacked rings with a staggered hexagonal pattern30 (Figure 2). This complex is predicted to recruit seven caspase-2 molecules and has a predicted molecular weight of 696.8 kDa, which matches the molecular weight of the PIDDosome complex produced upon temperature shift of extracts.18

A model of the PIDDosome for caspase-2 activation. The schematic model is based on the crystal structure of the PIDD-DD:RAIDD-DD core complex, which forms the center rings mediating oligomerization. The RAIDD CARD occupies the outer part of the PIDDosome bringing seven caspase-2 molecules into proximity facilitating dimerization. Adapted from Park et al30
PIDD Acts as a Molecular Switch for Survival and Apoptosis
PIDD, as well as activating caspase-2, can activate NFκB in response to DNA damage. In a further refinement of the PIDDosome model, Tschopp et al demonstrated that PIDD forms a separate complex that lacks RAIDD and caspase-2 but contains RIPK1 and NEMO (NFκB essential modulator)/IKKγ, which is the regulatory subunit of the inhibitor of κB (IκB) kinase complex.31 Recruitment of NEMO to the complex results in sumoylation of NEMO. Sumoylation is a post-translational modification that covalently attaches SUMO-1 (small ubiquitin-like modifier 1) to the target protein. There are a number of important potential effects of sumoylation including altering binding properties of the modified protein or retargeting the modified protein to a particular subcellular compartment.32 In the case of NEMO, sumoylation appears to be required to facilitate ubiquitination and phosphorylation of NEMO.33 Ubiquitination and phosphorylation of NEMO are dependent on the nuclear kinase ATM (ataxia telangiectasia mutated) while sumoylation is ATM-independent. These modifications shuttle NEMO from the nucleus to the cytoplasm where it induces the release of the inhibitor IκB from NFκB.33 NFκB then translocates to the nucleus to induce transcription of its many target genes, including numerous cytokines and other survival genes. Tschopp demonstrated that, in response to genotoxic stress, PIDD translocates to the nucleus where it assembles the PIDD–RIPK1–NEMO complex.31 This complex increases the amount of sumoylated NEMO in the nucleus, which in turn leads to an increased activation of NFκB. It is unclear how PIDD increases sumoylated NEMO as PIDD does not appear to be a direct SUMO ligase for NEMO. It has been hypothesized that PIDD can prevent desumoylation of NEMO,34 however, this has not been shown directly.
The recruitment of RAIDD and RIPK1 to the PIDDosome appears to be sequential. PIDD first recruits RIPK1 and NEMO, followed by RAIDD and caspase-2 at a later time point. Furthermore, in cells that are deficient in RIPK1, caspase-2 processing was observed to increase in response to DNA damage. Conversely, knockdown of RAIDD resulted in stronger SUMO modification of NEMO and increased NFκB activity in response to genotoxic stress.31 Thus, RAIDD and RIPK1 appear to compete for PIDD binding thus blocking the prosurvival or pro-apoptotic function of PIDD, respectively.
This bifurcation of function is purportedly due to the autoprocessing of PIDD. PIDD is processed by an intein-like mechanism between the two ZU5 domains at S446 and also at S588. PIDD autoprocessing produces three fragments: N-PIDD (48 kD) and PIDD-C (51 kD) are produced by cleavage at S446, while further processing at S588 produces the PIDD-CC fragment (37 kD). RAIDD binds PIDD-CC but not PIDD-C, and PIDD-CC is exclusively cytoplasmic. PIDD-C, in contrast, translocates to the nucleus and RIPK1 recruits only the PIDD-C fragment (Figure 3).27

Model of PIDD maturation and complex assembly. PIDD is cleaved at S446 to generate PIDD-N and PIDD-C. PIDD-C translocates to the nucleus where it recruites RIPK1 and NEMO, which triggers NFκB activation. Additional processing of PIDD at S588 produces PIDD-CC, which binds to RAIDD in the cytoplasm, which recruits and activates caspase-2. Adapted from Tinel et al27
The two fragments are formed in a sequential manner – first PIDD-C and then PIDD-CC. Formation of these fragments and consequent assembly of the RIPK1- and RAIDD-containing complexes appears to occur in a dose-dependent manner. In response to low levels of DNA damage, PIDD-C is produced and engages RIP and NEMO to activate NFκB-promoting cell survival. If the damage is too great, however, RAIDD and caspase-2 are recruited and the apoptotic program is engaged.27 Thus, PIDD may act as a molecular switch between death and survival pathways.
The Big Question: is the Function Apoptotic?
‘PIDDosome-based activation of caspase-2 is not toxic, and cells survive even if a notable fraction of the caspase-2 pool is activated. A second signal apparently is required for full commitment to caspase-2-mediated apoptosis.18’
This observation was made by Tschopp when he first described the PIDDosome. It represents a very important facet of the PIDD-caspase-2 pathway that unfortunately is often overlooked. This tells us that PIDD activation and more importantly caspase-2 activation is not always synonymous with induction of apoptosis and there is likely a threshold of caspase-2 activation that must be reached before caspase-2-dependent apoptosis is engaged. Emerging evidence supports this idea suggesting that caspase-2 may have functions in other cellular events such as regulation of cell cycle progression (see below).
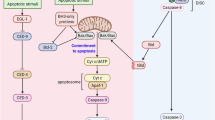
Caspase-2 has been shown to cleave the pro-apoptotic Bcl2 family member Bid.35 This is generally considered to be the mode through which caspase-2 initiates apoptosis. Thus, cleavage of Bid results in release of cytochrome c from the mitochondria to induce apoptosis (Figure 4). However, the ability of caspase-2 to cleave Bid is much more inefficient when compared with caspase-8, which can also engage the mitochondrial pathway through Bid cleavage. In vitro comparisons of caspase-2 and caspase-8 suggest that caspase-2 has approximately one-fourth the activity of caspase-8 toward Bid.36 Although it has been demonstrated that miniscule amounts of cleaved Bid are sufficient to induce cytochrome c release,37 low levels of active caspase-2 still may not produce enough active Bid to commit a cell to apoptosis in the absence of additional pro-apoptotic signals.

Caspase-2 pathways to apoptosis and cell cycle arrest. In response to cellular stresses PIDD activates caspase-2. Active caspase-2 cleaves Bid to form tBid, which induces mitochondrial outer membrane permeabilization. Active caspase-2 also cleaves MDM2 to form MDM2 p60, which binds to and stabilizes p53. p53 induces PIDD expression resulting in a feed-forward loop. Caspase-2 is also activated by ATM/ATR independently of p53 when Chk1 activity is repressed
A number of additional substrates for caspase-2 have been proposed, including golgin-160, CUX1 and ICAD (38, 39, 40 for a full list of substrates see 41), but most of these have only been shown in vitro and have not been verified in an in vivo situation, and the functional consequences of their cleavage by caspase-2 is unclear. It is likely that additional downstream targets of caspase-2 exist that when cleaved can either promote apoptosis or regulate additional functions of caspase-2.
The subcellular distribution of caspase-2 may also help inform how caspase-2 functions. Endogenous caspase-2 is localized throughout the cell but appears to be concentrated at the Golgi apparatus and the nucleus.38, 42 Results from our lab show that caspase-2 is activated in the cytosol in response to various stresses including heat shock, cytoskeletal disruption and DNA damage, as measured by bimolecular fluorescence complementation.43 It is therefore unclear what the role of caspase-2 in the nucleus is. It remains possible that caspase-2 can be activated in the nucleus but only in response to conditions that do not normally result in apoptosis, and in this scenario caspase-2 regulates other cellular events. Alternatively, caspase-2 may have a nuclear function that is independent of its enzymatic activity. These are open questions that require further exploration, which will likely uncover additional functions of this protease.
If caspase-2 does indeed function in cellular events distinct from apoptosis, this may explain the lack of an overt phenotype in mice that are deficient for caspase-2 and the caspase-2-related proteins PIDD and RAIDD. While caspase-8 and caspase-9 knockouts are embryonic lethal, caspase-2-deficient mice are born at expected Mendelian frequencies and the only measurable defect appears to be an excess of oocytes, although it has not been reported if this phenomenon is also observed in an independently generated caspase-2-deficient mouse line.42, 44 This argues against an essential role for caspase-2 in embryonic development. Similarly, PIDD-deficient animals and RAIDD-deficient animals are developmentally normal.17, 45, 46 In addition, there are very few cases where cells from caspase-2, RAIDD- and PIDD-deficient animals are profoundly resistant to apoptosis. Various independent studies have shown that thymocytes from caspase-2-deficient, RAIDD-deficient and PIDD-deficient mice are as sensitive as wild-type cells are to various cytotoxic agents, including γ-irradiation, etoposide or TNFα.17, 42, 45, 46
Some exceptions exist. Caspase-2-deficient and RAIDD-deficient splenocytes have been shown to be resistant to heat shock-induced death,47 but studies in other cell types such as Jurkat fail to see a difference in susceptibility to heat shock in the absence of caspase-2.48 Caspase-2-deficient embryonic fibroblasts have been shown to be significantly resistant to the cytoskeletal disruptors vincristine and paclitaxel/taxol but the latter is not phenocopied in PIDD-deficient cells.45, 49, 50 Caspase-2-deficient embryonic fibroblasts that were transformed with the oncogenes E1A and Ras were observed to be robustly resistant to γ-irradiation while the primary cells were not, again suggesting that caspase-2-induced apoptosis can be cell-type and context dependent.50 Finally, RAIDD-deficient fibroblasts are profoundly resistant to apoptosis induced by PIDD overexpression.17 However, it is important to note that caspase-2 activation has been observed to some extent in response to each of the stimuli listed above even in cases where apoptosis does not occur.18, 43, 47
Caspase-2 in Cell Division
Taken together this evidence suggests that, although activated caspase-2 can induce apoptosis under some circumstances, its activation does not mandatorily induce apoptosis and it may have other regulatory functions. Caspase-2-deficient embryonic fibroblasts reportedly proliferate at higher rates and PIDD overexpression induces growth suppression that is entirely dependent on RAIDD and partially dependent on caspase-2, indicating a potential role for caspase-2 in cell division.17, 50 In line with these observations, γ-irradiation-induced cell cycle arrest was shown to be profoundly impaired in the absence of caspase-2, indicating that caspase-2 may contribute to a cell cycle checkpoint.49
The mechanism of caspase-2-associated cell cycle arrest is unclear. Bid has also been shown to be associated with cell cycle arrest in response to DNA damage.51 However, a subsequent study failed to reproduce these findings, and therefore it is uncertain if Bid has any role in the regulation of cell cycle.52 A requirement for Bid in cell cycle arrest associated with caspase-2 has not been explored, but based on the latter results, it is unlikely that caspase-2-mediated proteolysis of Bid has a major role. Other targets of caspase-2 that carry out this function may exist.
Caspase-2 activity appears to be under tight regulation during the cell cycle and is repressed during normal mitosis. This repression is achieved by phosphorylation of serine residue 340, which is located in the interdomain linker between the large and small subunits of caspase-2.53 The mitosis-promoting kinase cdk1-cyclin B1 phosphorylates caspase-2 at this residue and the phosphatase PP1 (protein phosphatase 1) facilitates its dephosphorylation during interphase. Impairment of caspase-2 phosphorylation at this site during mitosis results in caspase-2 activation and cell death. PP1 is usually suppressed during mitosis, preventing dephosphorylation of caspase-2, but if the suppression becomes alleviated, in response to DNA damage perhaps, then the prediction is that caspase-2 activation would result. This may represent a damage limitation function of caspase-2; if the damage can be repaired then caspase-2 induces cell cycle arrest but in conditions of irreparable damage caspase-2 engages the apoptotic pathway.
Obviously, this type of checkpoint function is highly reminiscent of p53, begging the question: is p53 involved in this process? A recent paper suggests that caspase-2 enhances p53 function through cleavage of Mdm2 (mammalian double minute 2). A caspase cleavage site was identified in Mdm2 that separates the p53-binding region from the RING domain that is responsible for p53 ubiquitination. Consequently, cleavage at this site produces a 60 kD fragment of Mdm2 that can bind to p53 but does not degrade it, resulting in an overall increase in p53 stability. This results in a positive feedback loop where p53 induces PIDD upregulation and PIDD, in turn, stabilizes p53 through activation of caspase-254 (Figure 4). This model claims to explain why exogenous expression of PIDD can induce growth arrest in cells that are wild type for p53 but not in cells where p53 is absent or mutated.54 It is possible that caspase-2-mediated cleavage of Mdm2 contributes to other functions of p53 such as apoptosis, senescence and autophagy. In neurons, deficiency in caspase-2 results in increased autophagy in response to oxidative stress.55 p53 has been shown to promote and inhibit autophagy,56 however, it has not been investigated if p53 has a role in caspase-2-associated inhibition of autophagy in neurons.
There have been many reports of caspase-2 function that is independent of p53 so it is likely that cleavage of Mdm2 is not the sole way caspase-2 can induce growth arrest. For example, DNA damage triggers an ATM/ATR pathway that has been shown to induce caspase-2-dependent apoptosis when checkpoint kinase 1 (Chk1) is suppressed and this pathway bypasses a requirement for p53.57 (Figure 4).
Caspase-2 in Cancer and Aging
The potential role of caspase-2 as a cell cycle checkpoint protein suggests that loss of caspase-2 would have profound effects on the genomic stability of the cell and on the susceptibility of the organism to tumors. Caspase-2 knockout mice do not develop early spontaneous tumors.44 However, loss of caspase-2 in mice has been shown to be associated with accelerated tumorigenesis driven by transgenic c-Myc on the μ-enhancer (Eμ-Myc).50 Consistent with this, caspase-2-deficient embryonic fibroblasts are more efficiently transformed than wild-type cells.50 These data suggest that caspase-2 may function as a tumor suppressor and may even have an important role in cancer prevention.
Data regarding caspase-2 in human tumor samples have been somewhat inconsistent. Caspase-2 maps to the q34-35 segment of human chromosome 7.58 The 7q region is frequently deleted or affected in hematological neoplasms.59 Chromosomal deletions such as this commonly lead to the loss of a tumor suppressor, yet specific deletion of caspase-2 has not been reported. However, decreased caspase-2 and RAIDD expression was observed in mantle cell lymphoma tumor samples.60 In addition, drug resistance in childhood forms of acute lymphoblastic leukemia (ALL) has been correlated with decreased levels of caspase-2.61 In contrast, increased levels of caspase-2 in acute myelogenous leukemia and adult ALL have been associated with decreased patient survival.62, 63 Finally, a search for somatic mutations in caspase-2 failed to find these in acute leukemia and a number of solid tumor samples, including breast carcinoma and non-small cell lung cancer.64 Although there is little direct evidence of genetic abberations of caspase-2 in human cancer, the possibility exists that inactivation of caspase-2 in tumors occurs through disruption of the pathway through mutation or improper regulation of a protein that regulates caspase-2 activity.
For example, we observed that the heat shock protein, Hsp90α, inhibits caspase-2 activation.43 Hsp90 is commonly upregulated or has increased activity in many human tumors65, 66 and may represent a mode of inactivating caspase-2 to promote tumor growth. A role for Hsp90 in caspase-2 regulation is supported by results from Tschopp's lab, which showed that Hsp90 binds PIDD.67 However, in contrast to our results, Tschopp found that Hsp90 is an effector of PIDDosome activation and maturation. Therefore, the precise nature of the association between Hsp90 and PIDD/caspase-2 and its role, if any, in regulating the tumor suppressor function of caspase-2 remains to be determined.
In addition to a potential role as a tumor suppressor, there appear to be additional physiological consequences of impaired caspase-2 activation. Caspase-2-deficient mice have features consistent with accelerated ageing.68 The mice do not survive as long; the maximum lifespan is 10% shorter in the absence of caspase-2. In addition, old caspase-2-deficient mice show many enhanced aging-related features, including increased loss of bone mass as a result of increased bone resorption, lower body fat content and impaired hair regrowth. A subsequent study showed that caspase-2 deficiency attenuated diabetes-induced bone marrow adiposity indicated by decreased PPARy, which is a marker for adipocyte differentiation, suggesting a potential role for caspsase-2 in adipocyte differentiation.69
Remaining Questions
As described here, work from Jürg Tschopp's group has vastly increased our understanding of how caspase-2 is activated and how caspase-2 functions. Nevertheless, there is still so much we do not know about this protein, namely how it participates in cancer and aging. Activation of caspase-2 is clearly under tight regulatory control both in the regulation of PIDD and PIDDosome assembly, and by additional regulatory factors such as casapse-2 phosphorylation. However, the full cohort of caspase-2 regulatory proteins and factors has probably not been identified yet. The publication of the PIDD knockout mouse indicated that the caspase-2-containing high molecular weight complex formed upon temperature shift still assembles in the absence of PIDD.45 This suggests that alternative caspase-2 activation platforms exist that are independent of PIDD or can compensate for the loss of PIDD.
Adding to the complexity of caspase-2 regulation, it appears that PIDD complexes that are independent of caspase-2 and RAIDD exist. Tschopp identified a nuclear PIDD complex that contains PCNA (proliferating cell nuclear antigen), an important component of DNA repair machinery.70 PCNA acts as a DNA sliding clamp that binds DNA polymerase and prevents its dissociation from the template DNA strand. The PIDD–PCNA complex is induced by UV stimulation and appears to facilitate the loading of PCNA onto template DNA. Interestingly, PIDD-deficient animals were more sensitive to UV-induced apoptosis in the epidermis. Therefore, PIDD can indirectly regulate apoptosis in a manner that is independent of caspase-2.
Closing Remarks
‘The interest in this caspase has been dampened due to the lack of an overt phenotype of caspase-2-deficient mice and limited knowledge of its mode of activation and downstream targets 18’
Jürg Tschopp made this statement in the introduction of his publication characterizing the PIDDosome.18 With this important discovery, he reignited the interest in this caspase and the flame has remained strong ever since. Although many facets of the caspase-2 signaling pathway remain a mystery, the discovery that it may act as a safeguard against cancer makes it clear that caspase-2 has many critical and unique roles in regulating cell fate.
Abbreviations
- ALL:
-
acute lymphoblastic leukemia
- APAF-1:
-
apoptotic protease-activating factor-1
- ATM:
-
ataxia telangiectasia mutated
- CARD:
-
caspase recruitment domain
- Chk1:
-
checkpoint kinase 1
- DD:
-
death domain
- IκB:
-
inhibitor of κB
- LRR:
-
leucine-rich repeat
- Mdm2:
-
mammalian double minute 2
- NALP, NACHT:
-
LRR and PYD domains-containing protein
- NEMO:
-
NFκB essential modulator
- NFκB:
-
nuclear factor κB
- NOD:
-
nucleotide-binding oligomerization domain
- PCNA:
-
proliferating cell nuclear antigen
- PIDD:
-
p53-induced protein with a death domain
- PP1:
-
protein phosphatase 1
- RAIDD:
-
RIP-associated ICH-1/CAD-3 homologous protein with a death domain
- RIPK1:
-
receptor interacting protein kinase 1
- SUMO:
-
small ubiquitin-like modifier
- TNF:
-
tumor necrosis factor
- TNFR1:
-
tumor necrosis factor receptor 1
- TRADD:
-
tumor necrosis factor receptor type 1-associated death domain protein
References
Creagh EM, Conroy H, Martin SJ . Caspase-activation pathways in apoptosis and immunity. Immunol Rev 2003; 193: 10–21.
Lamkanfi M, Declercq W, Kalai M, Saelens X, Vandenabeele P . Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ 2002; 9: 358–361.
Krumschnabel G, Sohm B, Bock F, Manzl C, Villunger A . The enigma of caspase-2: the laymen′s view. Cell Death Differ 2009; 16: 195–207.
Vakifahmetoglu-Norberg H, Zhivotovsky B . The unpredictable caspase-2: what can it do? Trends Cell Biol 2010; 20: 150–159.
Boatright KM, Salvesen GS . Mechanisms of caspase activation. Curr Opin Cell Biol 2003; 15: 725–731.
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM et al. A unified model for apical caspase activation. Mol Cell 2003; 11: 529–541.
Green DR . Apoptotic pathways: ten minutes to dead. Cell 2005; 121: 671–674.
Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D et al. Substrate specificities of caspase family proteases. J Biol Chem 1997; 272: 9677–9682.
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 1997; 272: 17907–17911.
Baliga BC, Read SH, Kumar S . The biochemical mechanism of caspase-2 activation. Cell Death Differ 2004; 11: 1234–1241.
Read SH, Baliga BC, Ekert PG, Vaux DL, Kumar S . A novel Apaf-1-independent putative caspase-2 activation complex. J Cell Biol 2002; 159: 739–745.
Xue D, Shaham S, Horvitz HR . The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev 1996; 10: 1073–1083.
Hofmann K, Bucher P, Tschopp J . The CARD domain: a new apoptotic signalling motif. Trends Biochem Sci 1997; 22: 155–156.
Bouchier-Hayes L, Martin SJ . CARD games in apoptosis and immunity. EMBO Rep 2002; 3: 616–621.
Duan H, Dixit VM . RAIDD is a new ‘death’ adaptor molecule. Nature 1997; 385: 86–89.
Chou JJ, Matsuo H, Duan H, Wagner G . Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell 1998; 94: 171–180.
Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R et al. Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc Natl Acad Sci USA 2005; 102: 14314–14320.
Tinel A, Tschopp J . The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004; 304: 843–846.
Lin Y, Ma W, Benchimol S . Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet 2000; 26: 122–127.
Telliez JB, Bean KM, Lin LL . LRDD, a novel leucine rich repeat and death domain containing protein. Biochim Biophys Acta 2000; 1478: 280–288.
Aravind L, Dixit VM, Koonin EV . The domains of death: evolution of the apoptosis machinery. Trends Biochem Sci 1999; 24: 47–53.
Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB . The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature 1997; 386: 838–842.
Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M . Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature 1997; 386: 833–838.
Kobe B, Deisenhofer J . The leucine-rich repeat: a versatile binding motif. Trends Biochem Sci 1994; 19: 415–421.
Petrilli V, Dostert C, Muruve DA, Tschopp J . The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol 2007; 19: 615–622.
Inohara N, Nunez G . NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol 2003; 3: 371–382.
Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B et al. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J 2007; 26: 197–208.
Hu Y, Ding L, Spencer DM, Nunez G . WD-40 repeat region regulates Apaf-1 self-association and procaspase-9 activation. J Biol Chem 1998; 273: 33489–33494.
Martinon F, Tschopp J . NLRs join TLRs as innate sensors of pathogens. Trends Immunol 2005; 26: 447–454.
Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 2007; 128: 533–546.
Janssens S, Tinel A, Lippens S, Tschopp J . PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005; 123: 1079–1092.
Seeler JS, Dejean A . Nuclear and unclear functions of SUMO. Nat Rev Mol Cell Biol 2003; 4: 690–699.
Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S . Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 2003; 115: 565–576.
Wu ZH, Mabb A, Miyamoto S . PIDD: a switch hitter. Cell 2005; 123: 980–982.
Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES . Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem 2002; 277: 13430–13437.
Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR, Newmeyer DD . Caspase-2-induced apoptosis requires bid cleavage: a physiological role for bid in heat shock-induced death. Mol Biol Cell 2006; 17: 2150–2157.
Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ . Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 2000; 290: 1761–1765.
Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA et al. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol 2000; 149: 603–612.
Truscott M, Denault JB, Goulet B, Leduy L, Salvesen GS, Nepveu A . Carboxyl-terminal proteolytic processing of CUX1 by a caspase enables transcriptional activation in proliferating cells. J Biol Chem 2007; 282: 30216–30226.
Dahal GR, Karki P, Thapa A, Shahnawaz M, Shin SY, Lee JS et al. Caspase-2 cleaves DNA fragmentation factor (DFF45)/inhibitor of caspase-activated DNase (ICAD). Arch Biochem Biophys 2007; 468: 134–139.
Kitevska T, Spencer DM, Hawkins CJ . Caspase-2: controversial killer or checkpoint controller? Apoptosis 2009; 14: 829–848.
O′Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL et al. Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ 2002; 9: 832–841.
Bouchier-Hayes L, Oberst A, McStay GP, Connell S, Tait SW, Dillon CP et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell 2009; 35: 830–840.
Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 1998; 12: 1304–1314.
Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F et al. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol 2009; 185: 291–303.
Kim IR, Murakami K, Chen NJ, Saibil SD, Matysiak-Zablocki E, Elford AR et al. DNA damage- and stress-induced apoptosis occurs independently of PIDD. Apoptosis 2009; 14: 1039–1049.
Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR . In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol 2006; 8: 72–77.
Shelton SN, Dillard CD, Robertson JD . Activation of caspase-9, but not caspase-2 or caspase-8, is essential for heat-induced apoptosis in Jurkat cells. J Biol Chem 2010; 285: 40525–40533.
Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S . Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 2008; 27: 3393–3404.
Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S . A tumor suppressor function for caspase-2. Proc Natl Acad Sci USA 2009; 106: 5336–5341.
Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L et al. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 2005; 122: 593–603.
Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell 2007; 129: 423–433.
Andersen JL, Johnson CE, Freel CD, Parrish AB, Day JL, Buchakjian MR et al. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. Embo J 2009; 28: 3216–3227.
Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell 2011; 43: 57–71.
Tiwari M, Lopez-Cruzan M, Morgan WW, Herman B . Loss of caspase-2-dependent apoptosis induces autophagy after mitochondrial oxidative stress in primary cultures of young adult cortical neurons. J Biol Chem 2011; 286: 8493–8506.
Vousden KH, Ryan KM . p53 and metabolism. Nat Rev Cancer 2009; 9: 691–700.
Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008; 133: 864–877.
Kumar S . Inhibition of apoptosis by the expression of antisense Nedd2. FEBS Lett 1995; 368: 69–72.
Mitelman F, Kaneko Y, Trent JM . Report of the committee on chromosome changes in neoplasia. Cytogenet Cell Genet 1990; 55: 358–386.
Hofmann WK, de Vos S, Tsukasaki K, Wachsman W, Pinkus GS, Said JW et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood 2001; 98: 787–794.
Holleman A, den Boer ML, Kazemier KM, Beverloo HB, von Bergh AR, Janka-Schaub GE et al. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood 2005; 106: 1817–1823.
Estrov Z, Thall PF, Talpaz M, Estey EH, Kantarjian HM, Andreeff M et al. Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia. Blood 1998; 92: 3090–3097.
Faderl S, Thall PF, Kantarjian HM, Talpaz M, Harris D, Van Q et al. Caspase 2 and caspase 3 as predictors of complete remission and survival in adults with acute lymphoblastic leukemia. Clin Cancer Res 1999; 5: 4041–4047.
Kim MS, Chung NG, Yoo NJ, Lee SH . Somatic mutation of proapoptotic caspase-2 gene is rare in acute leukemias and common solid cancers. Eur J Haematol 2011; 86: 449–450.
Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003; 425: 407–410.
Bagatell R, Whitesell L . Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther 2004; 3: 1021–1030.
Tinel A, Eckert MJ, Logette E, Lippens S, Janssens S, Jaccard B et al. Regulation of PIDD auto-proteolysis and activity by the molecular chaperone Hsp90. Cell Death Differ 2011; 18: 506–515.
Zhang Y, Padalecki SS, Chaudhuri AR, De Waal E, Goins BA, Grubbs B et al. Caspase-2 deficiency enhances aging-related traits in mice. Mech Ageing Dev 2007; 128: 213–221.
Coe LM, Lippner D, Perez GI, McCabe LR . Caspase 2 deficiency protects mice from diabetes-induced marrow adiposity. J Cell Biochem 2011; 112: 2403–2411.
Logette E, Schuepbach-Mallepell S, Eckert MJ, Leo XH, Jaccard B, Manzl C et al. PIDD orchestrates translesion DNA synthesis in response to UV irradiation. Cell Death Differ 2011; 18: 1036–1045.
Acknowledgements
It is with immeasurable sadness that we offer this small testimony to the life and a few of the myriad contributions of our friend, Professor Jürg Tschopp. Jürg was a valued colleague, great friend and a brilliant scientist. One other small contribution is noted in another field, that of skiing; Dr Tschopp was the first to force one of us (DRG) to navigate the treacherous double-black diamond run, called ‘The Devil′s Crotch’ on Breckenridge Mountain (Jürg skied it effortlessly, while DRG did it significantly less so, but the latter has skied it since with fond memories). We will miss you in so many ways, Jürg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Bouchier-Hayes, L., Green, D. Caspase-2: the orphan caspase. Cell Death Differ 19, 51–57 (2012). https://doi.org/10.1038/cdd.2011.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.157
Keywords
This article is cited by
-
BID expression determines the apoptotic fate of cancer cells after abrogation of the spindle assembly checkpoint by AURKB or TTK inhibitors
Molecular Cancer (2023)
-
The caspase-2 substrate p54nrb exhibits a multifaceted role in tumor cell death susceptibility via gene regulatory functions
Cell Death & Disease (2022)
-
Caspase-2 regulates S-phase cell cycle events to protect from DNA damage accumulation independent of apoptosis
Oncogene (2022)
-
Effects of intravitreal injection of siRNA against caspase-2 on retinal and optic nerve degeneration in air blast induced ocular trauma
Scientific Reports (2021)
-
Uncovering the PIDDosome and caspase-2 as regulators of organogenesis and cellular differentiation
Cell Death & Differentiation (2020)
