
Abstract
Recently, the transcription factor encoded by tumor suppressor gene p53 was shown to regulate the expression of microRNAs. The most significant induction by p53 was observed for the microRNAs miR-34a and miR-34b/c, which turned out to be direct p53 target genes. Ectopic miR-34 expression induces apoptosis, cell-cycle arrest or senescence. In many tumor types the promoters of the miR-34a and the miR-34b/c genes are subject to inactivation by CpG methylation. MiR-34a resides on 1p36 and is commonly deleted in neuroblastomas. Furthermore, the loss of miR-34 expression has been linked to resistance against apoptosis induced by p53 activating agents used in chemotherapy. In this review, the evidence for a role of miR-34a and miR-34b/c in the apoptotic response of normal and tumor cells is surveyed.
Similar content being viewed by others

Main
Mutations in the p53 tumor suppressor gene are found in nearly all types of cancers.1, 2 The transcription factor encoded by the p53 tumor suppressor gene is post-transcriptionally activated by DNA damaging agents/radiation, oxidative stress or activation of oncogenes.3, 4, 5 The critical signals induced by these events are presumably DNA double-strand (ds) breaks, which activate ATM kinase that in turn phosphorylates p53. A number of additional modifications promote accumulation and increased transcriptional activity of p53, which directly regulates numerous target genes that mediate its diverse tumor suppressive effects.6 The induction of cell-cycle arrest, which can be transient or permanent (senescence), and the promotion of apoptosis in cases in which the damage is too severe are considered to be important for p53-mediated tumor suppression.5 The decision between these outcomes is determined by the level of p53 protein accumulation, with lower levels favoring arrest and higher levels promoting apoptosis.7 This effect is presumably because of differential affinities of p53-binding sites in the vicinity of target genes with pro arrest or cell death functions. Furthermore, proteins associating with p53 at specific promoters may influence this decision.7 Traditionally, the cellular effects of p53 are thought to be mediated by its ability to transactivate genes, which encode effector proteins that induce cellular processes: examples (and the phenotypic end points) are p21 (G1-arrest), 14-3-3σ (G2-arrest) and Puma (apoptosis).8, 9, 10 However, p53 has also been reported to induce the downregulation of specific proteins: for example, the p53-mediated loss of cyclin-dependent kinases (CDK4) and cyclins (Cyclin E2) may contribute to p53-induced cell-cycle arrest.11 Direct repressive effects of p53 on gene expression are mediated by binding to response elements that overlap with activating sites, by squelching of transcriptional activators and by recruitment of histone deacetylases.6, 12 Furthermore, p53 indirectly represses genes through activation of p21 that leads to association of pRB with E2F and therefore silencing of E2F target genes.13 For example, the survival factor, Bcl-2, is transcriptionally repressed by p53 through steric interference with DNA binding of the POU4F1 transcription factor at the Bcl-2 promoter.14
The discovery of microRNAs (miRNAs) suggested that the p53-mediated induction of miRNAs might contribute to the downregulation of proteins observed after p53 activation. miRNAs form a class of endogenously expressed, small noncoding RNAs that mediate post-transcriptional regulation of gene expression (reviewed in Ref.15, 16, 17, 18, 19). miRNA-encoding genes are transcribed by RNA polymerases II to yield primary transcripts (pri-miRNAs), which are processed by the nuclear RNase III enzyme, Drosha, to form stem-loop-structured miRNA precursor molecules. These resulting pre-miRNAs are transported to the cytoplasm in which the RNase III enzyme, Dicer, cleaves off the ds portion of the hairpin and generates a short-lived dsRNA of about 20–25 nucleotides in size. The duplex is subsequently unwound and only one strand gives rise to the mature miRNA, which is incorporated into miRNA–protein complexes (miRNPs). miRNAs guide miRNPs to partially complementary binding sites located in the 3′ untranslated region (3′-UTR) of the target mRNAs, which match to the seed sequences of the miRNAs. The bound miRNPs inhibit translation or destabilize the target mRNAs.20, 21 Both processes result in the downregulation of the protein encoded by the mRNA. Estimates based on bioinformatics as well as microarray analyses suggest that ∼30% of all genes are subject to regulation by multiple miRNAs.22
Roles of miRNAs in cancer
miRNAs have been implicated in the regulation of processes that are deregulated in cancer cells, as proliferation, differentiation and apoptosis.18 Alterations in miRNA expression in cancer have been documented in numerous studies and suggest that miRNAs critically contribute to the characteristics of cancer cells (for reviews see Refs.23, 24, 25, 26, 27). Furthermore, some miRNA-encoding genes have been classified as oncogenic or tumor suppressive genes according to their function in cellular transformation and altered expression in tumors. Tumor suppressive miRNAs may function by downregulating the products of proto-oncogenes. For example, the miRNA family let-7 targets expression of the oncogenes KRAS, NRAS and HMGA2 and its expression is diminished in lung tumors.28, 29, 30 Furthermore, the region encoding miR-15 and miR-16 is deleted in 65% of chronic lymphocytic leukemia (CLL) and in other tumors.26 As miR-15/16 targets the anti-apoptotic factor, Bcl-2, the loss of miR-15/16 may explain the upregulation of Bcl-2 in these tumors.
Connecting p53 and the miR-34 family
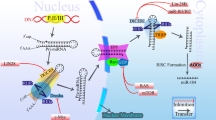
In 2007, reports from several laboratories showed that members of the miR-34 family are direct p53 targets, and their upregulation induces apoptosis and cell-cycle arrest (Figure 1).31, 32, 33, 34, 35, 36 In mammalians, the miR-34 family comprises three processed miRNAs that are encoded by two different genes: miR-34a is encoded by its own transcript, whereas miR-34b and miR-34c share a common primary transcript (Figure 2a). In mice, miR-34a is ubiquitously expressed with the highest expression in brain,38 whereas miR-34b/c is mainly expressed in lung tissues.35 These analyses also showed that miR-34a is expressed at higher levels than miR-34b/c, with the exception of the lung, in which miR-34b/c is dominantly expressed. Therefore, the two miR-34 genes presumably have tissue-specific functions. Similar to other p53-target genes, miR-34 genes may be the important targets for other signaling pathways involved in normal development. Whether this is the case, remains to be determined by genetic analysis in mice.

The miR-34 family as mediator of tumor suppression by p53. After the generation of double-strand breaks p53 is activated through ATM-kinases and transactivates target genes through consensus binding sites. The primary transcripts of the induced miR-34 genes are processed by DROSHA and DICER complexes. The mature miRNA is incorporated in the RISC complex and mediates inhibition of translation or RNA degradation of the indicated validated and presumably many others not yet confirmed targets. Exemplary cellular outcomes are indicated. Adapted with modifications from Ref37

Comparison of the miR-34 family members, miR-34a and miR-34b/c. (a) Structure of genomic loci of the human miR-34a and miR-34b/c genes. White and black boxes represent exons and miRNA hairpins, respectively. Hatched boxes indicate p53-binding sites; CpG islands are represented by thick black lines. The model is not shown to scale. Chromosomal (Chr) locations of the genes are provided. (b) Sequence alignment of the mature miR-34a, miR-34b and miR-34c molecules. The seed sequences are highlighted by gray shading. Asterisks indicate identical nucleotides
Functions of p53-induced miR-34 genes
Unexpectedly, the ectopic expression of miR-34 genes had rather drastic effects on cell proliferation and survival. Ectopic miR-34a and miR-34b/c caused a cell-cycle arrest in the G1 phase.32, 34, 35 In addition, miR-34b/c inhibited proliferation and colony formation in soft agar.36 Interestingly, the introduction of miR-34a and miR-34b/c into primary human diploid fibroblasts induced cellular senescence,32 a permanent form of cell-cycle arrest, which is presumably also relevant for organismal aging.39 Furthermore, re-expression of miR-34a induced apoptosis.31, 33, 34, 40 As cell-cycle arrest and apoptosis are common end points of p53 activation, miR-34 genes may be the potent mediators of tumor suppression by p53. Microarray analyses after ectopic introduction of different members of the miR-34 family into various cell lines revealed hundreds of putative, downregulated miR-34 targets.31, 32, 35 Interestingly, mRNAs with functions in the cell-cycle control and the DNA damage response were overrepresented among the transcripts downregulated by miR-34. Furthermore, the downregulated mRNAs showed an enrichment of miR-34 seed-matching sequences in their 3′-UTRs. Examples include CDK4/6, Cyclin E2, MET and Bcl-2.32, 35 For a survey of currently confirmed targets see Table 1. The observed downregulation of these proteins by miR-34a is presumably direct, because reporters carrying the 3′-UTR of the respective genes were inhibited by co-transfection of miR-34a and/or miR-34b/c in a manner dependent on the presence of an intact seed-sequence matching motif (Table 1). The high similarity among the three processed miR-34 family members (Figure 2b) suggested that they may have the same targets. Indeed, an expression analysis after separate transfection of miR-34a, miR-34b and miR-34c showed that the affected mRNAs were almost identical.32 However, differences in the affinities for targets may exist between the three miR-34 members, as perfect matches are only possible between certain miR-34 species and the 3′-UTR. An example is c-MYC, which seems to be regulated mainly by miR-34b/c.53, 54, 46 This can presumably be explained by the enhanced complementarity between the miR-34b seed sequence and the seed-matching sequence in the c-MYC 3′-UTR, when compared with miR-34a (Table 1; Figure 2b).
The induction of miR-34 genes allows p53 to regulate the expression of a large number of proteins, even after their transcripts have already been synthesized. This type of regulation may be advantageous in situations of cellular stress as it does not require the translation of additional effector proteins that would presumably take too long to allow time for repair. Furthermore, it facilitates the simultaneous regulation of numerous processes by p53. Interestingly, the mechanism of RNA interference has been implicated in other forms of stress response (reviewed in Ref.58). Furthermore, targeting of p53-induced mRNAs by miR-34 may contribute to the fine tuning of the p53 response and prevent an uncontrolled, irreversible response to p53 activation.59 The several modes of regulation exerted by the miRNAs were recently summarized:19 besides acting as an on/off switch the transcriptional induction of miRNAs may allow to fine tune protein levels to make cells more responsive to external signals. Furthermore, mRNAs that are transcriptionally repressed may be simultaneously targeted by miRNAs. This dual regulation allows an accelerated transition to the off-state. This may be the case for Bcl-2, because its transcription is directly repressed by p53, as discussed above,14 and its 3′-UTR is targeted by miR-34a.35
Inactivation of miR-34 in cancer
As cell-cycle arrest, senescence and apoptosis are tumor suppressive mechanisms, the permanent inactivation of members of the miR-34 family, which induce these cellular responses, may be a selective advantage for cancer cells. Besides decreased expression of miR-34 because of the inactivating mutations of p53 or the expression of viral inhibitors of p53, the miR-34-encoding genes themselves may be targets for the mutational or the epigenetic inactivation in cancer. Interestingly, miR-34a resides on the chromosomal locus 1p36, which has been proposed to harbor a tumor suppressor gene because it displays homozygous deletions in neuroblastoma and in other tumor types. An unbiased screen for genes with tumor suppressive function on 1p36 also revealed miR-34a as a candidate tumor suppressor gene.41 Interestingly, N-MYC, which is deregulated in the neuroblastoma, is a direct target of miR-34a.55 The correlation between 1p36 loss and miR-34a downregulation in the neuroblastoma was confirmed in this study. Furthermore, the expression of miR-34a was low or undetectable in 11 of 15 pancreatic cancer cell lines31 and the expression level of miR-34b was decreased by more than 90% in 6 out of 14 non-small cell lung cancer.35 However, this loss of expression did not strictly correlate with LOH, p53 mutation or increased CpG methylation.35 More recently, the epigenetic inactivation of miR-34a was identified in cell lines derived from some of the most common tumors (breast, lung, colon, kidney, bladder, pancreatic cancer and melanoma) and also in primary melanoma.38 In addition, CpG methylation of miR-34b/c was found in colorectal cancer,45 in oral squamous cell carcinoma60 and in malignant melanoma in which it correlated with metastatic potential.46 Furthermore, experimental animal models of liver carcinogenesis showed downregulation of miR-34a.61 Taken together, inactivation of the miR-34a and miR-34b/c genes presumably is a common event during tumorigenesis.
miR-34 and apoptosis
Before miR-34a was identified as a p53 target Welch et al.40 reported that ectopic miR-34a induces apoptosis when re-introduced into the neuroblastoma cell lines, which show decreased expression of miR-34a. In connection with the identification of miR-34a as a p53 target this finding was confirmed and extended. In cancer, miR-34-mediated apoptosis may be suppressed by inactivation of p53 and/or miR-34 genes (Figure 3). Chang et al.31 showed that miR-34a-induced apoptosis is at least, in part, dependent on the presence of wild-type p53 indicating that miR-34a may feed back to p53 (see below). Furthermore, locked nucleic acids directed against miR-34a protect cells to some extent from the DNA damage-induced apoptosis in wild-type p53-expressing cells.33 Bommer et al.35 showed that Bcl-2 is targeted by miR-34a and that miR-34a-defective MEFs show a decrease in spontaneous apoptosis. Whether miR-34 induces apoptosis presumably depends on the cellular context and therefore the expression levels of the respective miR-34 target proteins involved in the regulation of apoptosis. For example, we observed apoptosis after induction of a conditional miR-34a allele in H1299 lung cancer cells, whereas the same construct induces a G1-arrest in U-2OS osteosarcoma cells (Hermeking et al., unpublished results and Ref.34). In addition, the level of miR-34 expression may affect the decision between apoptosis and cell-cycle arrest.

The p53/miR-34 pathway regulates apoptosis and is altered in cancer. As described in the text, SIRT1 is repressed by miR-34a. As a result, p53 deacetylation by SIRT1 is decreased and leads to increased transcription of p53 targets, such as PUMA. Together with the downregulation of Bcl-2 and other anti-apoptotic proteins (NN) miR-34 activation promotes apoptosis. In cancer, the p53-miR-34 connection is often targeted by the indicated alterations. As a result, the induction of apoptosis is diminished after the DNA damage induced by chemotherapy
miR-34a feeds back to p53
miR-34a was shown to target SIRT1 mRNA leading to translational repression of SIRT1 (Figure 3).56 SIRT1 is an NAD-dependent deacetylase, which has been shown to inhibit several pro-apoptotic proteins.62 As mentioned above, apoptosis induced by the re-introduction of miR-34a is dependent on p53 to some extent.31 This observation was confirmed by Yamakuchi et al.56 and linked to the targeting of SIRT1 mRNA by miR-34a. They could show that p53 acetylation on lysine 382 increases after miR-34a expression. This was associated with increased transcriptional activity of p53, which led to the induction of p21 and PUMA. The latter presumably mediated apoptosis in the scenario. Ectopic expression of a miR-34a-resistant SIRT1 cDNA partially rescued miR-34a-induced apoptosis indicating that additional anti-apoptotic proteins may be targeted by miR-34a. In summary, the regulation of SIRT1 by miR-34a is part of a positive feedback loop that leads to further activation of p53, once it has been activated (Figure 3). As SIRT1 activity is NAD-dependent the metabolic state of the cell may also influence the effectiveness of this regulation. Loss of miR-34 through genetic or epigenetic mechanisms interrupts this feedback resulting in lower p53 activity and thereby provides a selective advantage for cancer cells. Fujita et al.42 also reported regulation of SIRT1 by miR-34a, although they exclusively observed the effects of miR-34a on the expression of SIRT1 mRNA and not on the translation. Another feedback loop between p53 and miR-34a may involve the downregulation of the p53-inhibitor HDMX by miR-34a.57
miR-34 in cancer therapy and detection
Given the tumor suppressive functions ascribed to the miR-34 family, it will be interesting to determine whether the detection of miR-34 expression also has diagnostic or prognostic potential in other tumor types. When compared with the generation of mRNA expression profiles the detection of miRNAs may have diagnostic advantages. For example Lu et al.63 could show that the expression analysis of 217 miRNAs is superior to genome-wide analysis of mRNAs for the purpose of classifying tumors.
Zenz et al.64 found that the expression of miR-34a is decreased in CLL. This was associated with p53 mutations, chemotherapy (fludarabine)-refractory disease, impaired DNA damage response and decreased apoptosis.64 Mraz et al.65 also found that miR-34a is consistently downregulated in CLL with p53 mutations. This implies that the detection of miR-34a expression may potentially be used as a predictor of therapy response. Furthermore, the restoration of miR-34a activity may be useful to prevent chemotherapy resistance. After the administration of the MDM2 inhibitor, Nutlin-3, to human diploid fibroblasts, induction of miR-34a and miR-34b/c, as well as senescence was observed.66 As mentioned above, the cellular context may determine the outcome of miR-34 induction. In the cancer cells expressing wild-type p53 induction of miR-34 expression by Nutlin-3 presumably leads to apoptosis. The identification of additional miR-34 targets regulating apoptosis may help to find further points of intervention.
Outlook
siRNAs have been proposed for clinical applications, and the feasibility of this approach is currently being tested.67 If specific introduction of siRNAs into tumors is successful, it may be possible to restore miR-34 function for cancer therapeutic purposes in the future. Furthermore, the possibility to detect epigenetic inactivation of miR-34a and/or miR-34b/c genes using methylation-specific PCR or loss of miR-34 expression holds cancer diagnostic and prognostic potential for the future.
Abbreviations
- ATM:
-
ataxia teleangiectasia mutated
- CDK:
-
cyclin-dependent kinase
- CLL:
-
chronic lymphocytic leukemia
- CpG:
-
cytosine–guanosine dinucleotide
- HDAC:
-
histone deacetylase
- LNA:
-
locked nucleic acids
- LOH:
-
loss of heterozygosity
- miRNA:
-
microRNA
- miRNP:
-
miRNA–protein complexes
- NSCLC:
-
non-small cell lung cancer
- siRNA:
-
small interfering RNA
- SIRT1:
-
silent information regulator 1
- UTR:
-
untranslated region of mRNA
References
Hollstein M, Sidransky D, Vogelstein B and Harris CC . p53 mutations in human cancers. Science 1991; 253: 49–53.
Soussi T . p53 alterations in human cancer: more questions than answers. Oncogene 2007; 26: 2145–2156.
Vogelstein B, Lane D and Levine AJ . Surfing the p53 network. Nature 2000; 408: 307–310.
Oren M . Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10: 431–442.
Vousden KH and Lane DP . p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8: 275–283.
Riley T, Sontag E, Chen P and Levine A . Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–412.
Aylon Y and Oren M . Living with p53, dying of p53. Cell 2007; 130: 597–600.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R and Trent JM et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–825.
Yu J, Zhang L, Hwang PM, Kinzler KW and Vogelstein B . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7: 673–682.
Hermeking H, Lengauer C, Polyak K, He TC, Zhang L and Thiagalingam S et al. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1997; 1: 3–11.
Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B and Meyn RE et al. Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. J Biol Chem 2006; 281: 25134–25142.
Ho J and Benchimol S . Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ 2003; 10: 404–408.
Lohr K, Moritz C, Contente A and Dobbelstein M . p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem 2003; 278: 32507–32516.
Budhram-Mahadeo V, Morris PJ, Smith MD, Midgley CA, Boxer LM and Latchman DS . p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J Biol Chem 1999; 274: 15237–15244.
Valencia-Sanchez MA, Liu J, Hannon GJ and Parker R . Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006; 20: 515–524.
Peters L and Meister G . Argonaute proteins: mediators of RNA silencing. Mol Cell 2007; 26: 611–623.
Chen K and Rajewsky N . The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 2007; 8: 93–103.
Kloosterman WP and Plasterk RH . The diverse functions of microRNAs in animal development and disease. Dev Cell 2006; 11: 441–450.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233.
Meister G and Tuschl T . Mechanisms of gene silencing by double-stranded RNA. Nature 2004; 431: 343–349.
Pillai RS, Bhattacharyya SN and Filipowicz W . Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol 2007; 17: 118–126.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM and Castle J et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005; 433: 769–773.
Garzon R, Fabbri M, Cimmino A, Calin GA and Croce CM . MicroRNA expression and function in cancer. Trends Mol Med 2006; 12: 580–587.
Cummins JM and Velculescu VE . Implications of micro-RNA profiling for cancer diagnosis. Oncogene 2006; 25: 6220–6227.
Esquela-Kerscher A and Slack FJ . Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–269.
Calin GA and Croce CM . MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–866.
Lee YS and Dutta A . MicroRNAs in cancer. Annu Rev Pathol 2009; 4: 199–227.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R and Cheng A et al. RAS is regulated by the let-7 microRNA family. Cell 2005; 120: 635–647.
Mayr C, Hemann MT and Bartel DP . Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007; 315: 1576–1579.
Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K and Yi M et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006; 9: 189–198.
Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M and Lee KH et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752.
He L, He X, Lim LP, de Stanchina E, Xuan Z and Liang Y et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134.
Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N and Moskovits N et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26: 731–743.
Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A and Menssen A et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007; 6: 1586–1593.
Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R and Love RE et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 2007; 17: 1298–1307.
Corney DC, Flesken-Nikitin A, Godwin AK, Wang W and Nikitin AY . MicroRNA-34b and microRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res 2007; 67: 8433–8438.
Hermeking H . p53 enters the microRNA world. Cancer Cell 2007; 12: 414–418.
Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T and Korner H et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008; 7: 2591–2600.
Collado M, Blasco MA and Serrano M . Cellular senescence in cancer and aging. Cell 2007; 130: 223–233.
Welch C, Chen Y and Stallings RL . MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007; 26: 5017–5022.
Cole KA, Attiyeh EF, Mosse YP, Laquaglia MJ, Diskin SJ and Brodeur GM et al. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol Cancer Res 2008; 6: 735–742.
Fujita Y, Kojima K, Hamada N, Ohhashi R, Akao Y and Nozawa Y et al. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun 2008; 377: 114–119.
Ji Q, Hao X, Meng Y, Zhang M, Desano J and Fan D et al. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008; 8: 266.
Sun F, Fu H, Liu Q, Tie Y, Zhu J and Xing R et al. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett 2008; 582: 1564–1568.
Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K and Shinomura Y et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68: 4123–4132.
Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M and Blanco D et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 2008; 105: 13556–13561.
Pigazzi M, Manara E, Baron E and Basso G . miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukemia. Cancer Res 2009; 69: 2471–2478.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP and Burge CB . Prediction of mammalian microRNA targets. Cell 2003; 115: 787–798.
Tazawa H, Tsuchiya N, Izumiya M and Nakagama H . Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 2007; 104: 15472–15477.
Li N, Fu H, Tie Y, Hu Z, Kong W and Wu Y et al. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett 2009; 275: 44–53.
Yan D, Zhou X, Chen X, Hu DN, Dong XD and Wang J et al. MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Invest Ophthalmol Vis Sci 2009; 50: 1559–1565.
Migliore C, Petrelli A, Ghiso E, Corso S, Capparuccia L and Eramo A et al. MicroRNAs impair MET-mediated invasive growth. Cancer Res 2008; 68: 10128–10136.
Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P and Bellan C et al. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol 2008; 216: 440–450.
Kong YW, Cannell IG, de Moor CH, Hill K, Garside PG and Hamilton TL et al. The mechanism of micro-RNA-mediated translation repression is determined by the promoter of the target gene. Proc Natl Acad Sci USA 2008; 105: 8866–8871.
Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT and Tsang P et al. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008; 27: 5204–5213.
Yamakuchi M, Ferlito M and Lowenstein CJ . miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA 2008; 105: 13421–13426.
Markey M and Berberich SJ . Full-length hdmX transcripts decrease following genotoxic stress. Oncogene 2008; 27: 6657–6666.
Leung AK and Sharp PA . microRNAs: a safeguard against turmoil? Cell 2007; 130: 581–585.
Cohen SM, Brennecke J and Stark A . Denoising feedback loops by thresholding—a new role for microRNAs. Genes Dev 2006; 20: 2769–2772.
Kozaki K, Imoto I, Mogi S, Omura K and Inazawa J . Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res 2008; 68: 2094–2105.
Tryndyak VP, Ross SA, Beland FA and Pogribny IP . Down-regulation of the microRNAs miR-34a, miR-127, and miR-200b in rat liver during hepatocarcinogenesis induced by a methyl-deficient diet. Mol Carcinog 2008; [E-pub ahead of print]. doi: 10.1002/mc.20484.
Brooks CL and Gu W . How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 2009; 9: 123–128.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J and Peck D et al. MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–838.
Zenz T, Mohr J, Eldering E, Kater AP, Buhler A and Kienle D et al. MiR-34a as part of the chemotherapy resistance network in chronic lymphocytic leukemia. Blood 2009; 113: 3801–3808.
Mraz M, Malinova K, Kotaskova J, Pavlova S, Tichy B and Malcikova J et al. miR-34a, miR-29c and miR-17-5p are downregulated in CLL patients with TP53 abnormalities. Leukemia 2009; [E-pub ahead of print]. doi: 10.1038.
Kumamoto K, Spillare EA, Fujita K, Horikawa I, Yamashita T and Appella E et al. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res 2008; 68: 3193–3203.
de Fougerolles A, Vornlocher HP, Maraganore J and Lieberman J . Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov 2007; 6: 443–453.
Acknowledgements
The author wishes to thank AK Kretzschmar for help with preparing figure 2 and B Hansen for compiling data for table 1. Work in Heiko Hermeking's lab is supported by the DFG, the Deutsche Krebshilfe and the Rudolf-Bartling-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
This article is cited by
-
Small noncoding RNAs and sperm nuclear basic proteins reflect the environmental impact on germ cells
Molecular Medicine (2024)
-
Profiling miRNAs in tear extracellular vesicles: a pilot study with implications for diagnosis of ocular diseases
Japanese Journal of Ophthalmology (2024)
-
Stress, microRNAs, and stress-related psychiatric disorders: an overview
Molecular Psychiatry (2023)
-
CRISPR/Cas9-mediated inactivation of miR-34a and miR-34b/c in HCT116 colorectal cancer cells: comprehensive characterization after exposure to 5-FU reveals EMT and autophagy as key processes regulated by miR-34
Cell Death & Differentiation (2023)
-
Retraction Note: Functional studies of miR-130a on the inhibitory pathways of apoptosis in patients with chronic myeloid leukemia
Cancer Gene Therapy (2023)
