
Abstract
Multiple sclerosis (MS) has been classically regarded as a disorder of the white matter of the central nervous system (CNS). However, early alterations of the neuronal compartment occurring in this disorder are partially independent of demyelination. Soluble inflammatory cytokines and glutamate have been proposed as major determinants of neurodegeneration in MS as well as in its experimental animal model, namely experimental autoimmune encephalomyelitis (EAE). The relationship between these two major determinants has been largely elusive. In recent years, unexpected connections have emerged between immune cells and soluble cytokines on the one hand, and synaptic transmission and neurodegeneration on the other. Neurophysiological recordings have recently shown that glutamate-mediated excitatory postsynaptic currents are enhanced during the early phase of EAE, because of altered expression and phosphorylation of AMPA receptors and the downregulation of the immediate early gene Arc/Arg3.1. The synaptic alterations occurring during neuroinflammatory diseases are largely mediated by inflammatory cytokines released from infiltrating T cells and from activated microglia, and are responsible, at least in part, for irreversible dendritic pathology. Collectively, the data covered in this review article suggest that CNS-confined inflammation in MS is associated with the release of soluble molecules, which are capable of altering excitatory synaptic transmission and, finally, of stimulating secondary neurodegenerative gray matter pathology.
Similar content being viewed by others
Main
In recent years, the classical dichotomy between inflammatory and degenerative diseases of the central nervous system (CNS) has been challenged by a series of discoveries showing that the two processes coexist both in degenerative disorders – such as Alzheimer's disease or amyotrophic lateral sclerosis – and in classical inflammatory disorders, such as multiple sclerosis (MS).1, 2
Multiple sclerosis is the most common cause of neurological disability in young adults, believed to follow immune-mediated aggression of the central myelin by blood-borne autoreactive T lymphocytes. In many cases, the clinical course of MS is characterized by recurrent episodes of neurological deficits in the early stages (relapsing-remitting MS (RRMS)), and by a progressive neurological decline after 15–20 years from the diagnosis (secondary progressive MS).3 A minority of MS patients has a progressive course from the onset, with rare or nonexistent clinical relapses (primary progressive MS).3, 4
Inflammatory demyelination of the white matter of the CNS is the primary pathological hallmark of RRMS, whereas gray matter atrophy and neuronal degeneration predominate in the progressive forms of the disease.5 Recent discoveries, however, have led to a reconsideration of the perceived relationship between inflammation and neurodegeneration in MS, because it is now accepted that one process is not simply the culminating event of the other. Inflammation and neurodegeneration, in fact, seem to be intermingled rather than occurring in series in this disorder. Common molecular pathways bringing together the two pathological processes have, in fact, been described.1, 2 Activated immune cells are able to damage neurons in the absence of any antigen specificity.6, 7 Damaged neurons do trigger local CNS-confined (auto)immune responses.8 Brain atrophy and cortical thinning, reduction in neuronal-specific markers, iron deposition and other gray matter abnormalities have been found in the inflammatory phase of MS (RRMS) subjects with conventional and advanced in vivo imaging techniques. Cognitive deficit has been described in a relevant percentage of MS patients already at the first episode of the disease (clinically isolated syndrome).9 All together, these pieces of evidence support the concept that gray matter and white matter alterations in MS patients might initially occur independently, as a result of CNS-confined inflammatory processes, and then evolve intermingled as neurodegeneration can be the consequence of primary neuroinflammation and vice versa.3, 4, 5 To support this working hypothesis, it is noteworthy to mention that in addition to focal axon damage secondary to demyelination, and to retrograde, anterograde and transneuronal degeneration, axon transection can be induced by cytotoxic T cells as well as soluble molecules released by CNS resident (microglia) and invading (blood-borne mononuclear cells) inflammatory cells (e.g., axon-specific antibodies, complement, NO, oxygen radicals, proteases and eicosanoids).10 Finally, neuronal damage in MS can also be due to acquired neuronal channelopathies, altered activity of sodium/calcium exchanger, glutamate-mediated excitotoxicity, intraneuronal calcium accumulation and inhibition of mitochondrial respiratory chain.10, 11
Role of Glutamate in MS Neuronal Injury
Glutamate is the major excitatory neurotransmitter in the CNS, and its concentration at the synaptic cleft is finely regulated by multiple mechanisms, which include the glutamate-glutamine cycle and the activity of glutamate uptake and transport mechanisms. Altered glutamate metabolism resulting in excessive synaptic excitation has been convincingly associated with neuronal damage in many acute and chronic neurological disorders, through a process termed excitotoxicity.12 Excitotoxic neuronal damage is therefore the result of abnormal intracellular ion accumulation flowing through NMDA and non-NMDA glutamate receptors, leading to membrane lysis and to the activation of fatal degenerative processes and dendritic apoptosis.12 Also, metabotropic glutamate (mGlu) receptors have been claimed to modulate excitotoxic neuronal damage, as group I mGlu receptors (mGlu 1 and 5) have a preferential synaptic localization and shape both AMPA13, 14 and NMDA signaling and toxicity,15 whereas group II mGlu (mGlu 2 and 3) and group III mGlu receptors (mGlu 4, 6, 8) are preferentially located in presynaptic neuronal elements or in glial cells, where they can be both neuroprotective15 or detrimental for neuronal survival.16, 17
Early studies have identified the neurotransmitter glutamate as an important determinant of neurodegenerative damage in the course of MS.18, 19 In fact, glutamate levels increase in the cerebrospinal fluid20, 21 and in the brains of MS patients,18, 19 whereas the AMPA, NMDA and kainate receptors are upregulated.22 In addition, expressions of glutamate transporters are altered in MS23, 24, 25 and in animal models of the disease,26, 27 and the loss of glutamate transporters in cortical lesions correlates with microglial activation and synaptic damage.28 Finally, overactivation of glutamate receptors causes MS-like lesions,29 whereas glutamate receptor antagonists exert beneficial effects in experimental autoimmune encephalomyelitis (EAE)30, 31, 32, 33 and in MS,34 by limiting not only oligodendrocyte but also neuronal damage.30, 31, 35 When administered at the onset of neurological deficits, various AMPA receptor antagonists (NBQX, MPQX, GYKI52466, GYKI53773) have been found to be capable of attenuating clinical decline, independently of any effect on CNS lymphocyte infiltration.18, 19 The same was true for the NMDA receptor antagonists memantine, amantadine and MK-801,27, 28 and for the inhibitor of glutamate transmission riluzole.36 We have recently found that the in vivo blockade of AMPA receptors not only ameliorated the clinical course of EAE, but also preserved dendritic spine loss occurring in this model of MS. These results support the involvement of AMPA receptors in EAE-induced dendritic spine degeneration.35
Furthermore, glutamate-triggered neurodegeneration might be favored by a parallel alteration of GABA transmission in MS brains, causing an imbalance between synaptic excitation and inhibition. Accordingly, a neurophysiological study has reported a marked brain hyperexcitability in MS patients, likely resulting from impaired GABA-mediated synaptic inhibition,37 and preliminary evidence is also available in mice with EAE (D. Centonze, manuscript in preparation).
Alteration of Synaptic Transmission in Experimental MS
Activated leukocytes and microglia are the most accredited source of glutamate during inflammation of the CNS,38, 39 whereas synaptic release has received some attention only recently. By means of conventional whole-cell patch-clamp recordings from single neurons, we recently addressed glutamate-mediated synaptic currents in corticostriatal brain slices from C57Bl/6 mice immunized with myelin oligodendrocyte glycoprotein 35–55 peptide to induce EAE.35 We found that the duration of glutamate-mediated spontaneous and miniature excitatory postsynaptic currents (sEPSCs and mEPSCs) was increased in striatal cells in both the presymptomatic and clinical phases of the disease, as expected for enhanced excitatory transmission in these neurons. In particular, we found a slower decay phase of sEPSC and mEPSCs, whereas rise time and amplitude of these synaptic events were normal. Glutamate NMDA receptors were not involved in this alteration, which was entirely mediated by AMPA receptors. As the activity of AMPA receptors is known to be regulated by both the expression and phosphorylation states of specific receptor subunits,40 we also measured the expression of the GluR1 subunit of AMPA receptors and its phosphorylation at the Ser845 residue. In isolated striatal postsynaptic membranes and in the whole dissected striatum, we found that both total GluR1 and GluR1-p-Ser845 expression were upregulated in EAE, whereas markers of presynaptic activity were normal.35
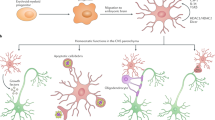
These results suggested to us that postsynaptic sites of striatal neurons were exposed to excessive glutamate activity in EAE, possibly causing synaptic degeneration since the very early phases of the disease, as schematically depicted in Figure 1. However, it is extremely unlikely that excessive AMPA receptor function is the only synaptic event leading to dendritic and neuronal degeneration in EAE brains, as many transmitter systems and receptor subtypes are altered in this disorder and in MS. Reduced signaling through cannabinoid CB1 receptors, for example, has been identified as an early synaptic change in EAE,41 and this alteration might well contribute to dendritic pathology and neuronal loss. In fact, we have recently found that voluntary exercise can attenuate the clinical deficits of mice with EAE by preserving cannabinoid CB1-mediated transmission and dendritic integrity even in the absence of overt effects on glutamate transmission. EAE-induced abnormalities of glutamate-mediated sEPSCs were, in fact, still present in mice with EAE with access to a running wheel, although this therapeutic environmental manipulation was capable of reducing clinical score and dendritic spine loss in these mice.42

Glutamate is released not only by excitatory synaptic terminals but also by infiltrating lymphocytes and by activated microglia in MS brains. Glutamate binds to and activates abnormally sensitive AMPA receptors, leading to synaptic and neuronal degeneration. Blockade of AMPA receptors preserves neuronal integrity and ameliorates the clinical course of mice with EAE
Along with AMPA and NMDA receptors, mGlu receptors are also likely to contribute to glutamate transmission changes in EAE and in MS brains. For example, a significant reduction in the expression of mGlu 1 receptors has been reported to occur concomitantly with increased mGlu 5 receptors in the cerebellum of mice with EAE as well of patients with MS,43 whereas both receptors increase in the context of MS lesions.23 Also, mGlu 2, 3, 4 and 8 have been found to increase in active MS lesions, especially in reactive astrocytic and microglial cells.23, 24 The impact of these complex adaptations on synaptic transmission and secondary neurodegeneration has not yet been explored, and it is difficult to predict, as both neuroprotective and detrimental effects could be postulated. Accordingly, mGlu 1 receptors are critically involved in AMPA-mediated long-term synaptic depression,13, 14 implying that the dramatic downregulation of these receptors seen in EAE and in MS might favor AMPA-mediated neuronal toxicity. In line with this hypothesis, treatment with the mGlu 1 receptor enhancer resulted in ameliorated motor performance in mice with EAE.43 Increased signaling through mGlu 5 receptors might also be involved in the neurodegenerative damage of MS, as the activation of these receptors has been found to exacerbate NMDA-mediated excitotoxicity.15
Role of EAE-Specific Inflammatory T Cells in Synaptic Functioning
Under physiological conditions, T-cell trafficking from peripheral tissues to the brain is essential for CNS immunosurveillance from danger signals. Such immunosurveillance may also include the preservation of an appropriate synaptic transmission, which is essential for the physiological functioning of the CNS cells. As a matter of fact, it has been recently shown that inflammatory effector T cells can inhibit hippocampal long-term potentiation (LTP) in vitro.44
In EAE, effector T cells activated in the periphery against myelin antigens and subsequently invading the CNS are considered as the primum movens of the experimental disease and, as shown by passive T-cell transfer experiments, do substantially contribute to clinical deficits that are caused by primary inflammation, followed by secondary demyelination and axonal damage.3 Several lines of evidence suggest that T-cell CNS invasion might also considered as the first step in the chain of events leading to brain inflammation and secondary neurodegeneration occurring in MS patients.39, 45 Thus, we first investigated whether EAE-specific T cells were involved in the abovementioned synaptic transmission deficits seen in EAE.
In the presymptomatic and symptomatic phases of EAE, we found inflammatory infiltrates in the striatum and the cortex mainly composed by CD3+ T cells. This indicates that not only the white matter but also the gray matter structures are infiltrated by these cells during experimental MS. After aseptically removing the spleens and lymph nodes from donor mice with EAE, we prepared suspensions of CD3+ T lymphocytes and placed them onto a single brain slice obtained from normal mice, to see whether the activated T lymphocytes were able to replicate the synaptic alterations seen in EAE brains. EAE-specific CD3+ T cells were indeed able to alter glutamate-mediated sEPSC kinetic properties in slices from control animals, in a way reminiscent of the defects seen in EAE brains. However, it was not only sEPSC decay time but also sEPSC rise time that was altered by T lymphocytes, possibly indicating that these inflammatory cells change glutamate-mediated synaptic transmission by altering the activity of both NMDA and AMPA receptors.35 T cells infiltrating the brain not only regulate synaptic transmission and plasticity mediated by glutamate,30, 35 but are also regulated by this neurotransmitter, as they respond to glutamate by activating a neuroprotective pathway.46 Thus, glutamate-mediated control over T cells invading the CNS might function as a feedback regulatory mechanism to limit T-cell-induced excitotoxic damage, but a failure of this potentially neuroprotective system has been described in T lymphocytes from MS patients.47
Role of Activated Microglia in EAE-Specific Synaptic Alteration
The identification of CNS-resident astroglia and microglia as important components of innate immunity was indeed crucial to understand the role of inflammation in neuronal damage. Although reactive gliosis was considered as a well-known event solely accompanying both acute and chronic neuronal damage epiphenomenally, it is now becoming clear that this phenomenon is also of pivotal importance in regulating and instructing the harmful inflammatory response causing neuronal network derangement. On the one hand, activated astroglia can mediate both the protective and toxic effects during neurodegenerative diseases.48 On the other, reactive microglia has an active role in the defensive attack against viruses and bacteria, but intense activation can also be detrimental for the survival of neighboring cells when this activation aims at clearing up apoptotic cells or neuron debris during neurodegenerative disorders.49, 50
This dual role of glial cells in the CNS during inflammatory and neurodegenerative disorders has prompted us to hypothesize that infiltrating CD3+ T cells might activate, through the release of proinflammatory cytokines, endogenous microglial cells, which in turn could contribute to the synaptic alterations we have observed in mice with EAE. To test this hypothesis, we stimulated microglial primary cell cultures with a cocktail of proinflammatory Th1 cytokines (tumor necrosis factor-α, TNFα; interferon-γ, IFNγ and interleukin-1β, IL-1β), which are released within the striatum during acute inflammation.35 We next tested whether or not activation of microglia in the striatal areas might lead to synaptic transmission alterations. Incubation of control brain slices with in vitro activated microglia increased the duration of sEPSCs by slowing their decay phases. These results mimicked the synaptic deficits seen in mice with EAE. We also found that activated microglia deposited on slices from mice with EAE did not further increase the sEPSC decay phase, indicating that EAE occluded the effects of activated microglia on this parameter.35 These results support the conclusion that microglia, activated by effector T cells invading the brain, are responsible for synaptic alterations in EAE (Figure 2).

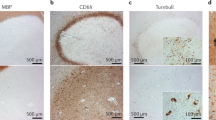
Excitatory synaptic transmission is altered in the striatum of mice with EAE. (a) Photograph (infrared videomicroscope, × 40) of a striatal neuron during a patch-clamp experiment. The recorded striatal neuron is marked by an orange circle. (b) Schematic representation of a single neuron electrophysiological recording in corticostriatal brain slice in a control mouse. The draws on the right are examples of AMPA receptor-mediated spontaneous excitatory postsynaptic currents (sEPSCs). (c–e) The decay phase of AMPA receptor-mediated sEPSCs is slower in mice with EAE (c) and in control slices incubated in the presence of activated microglial cells (d). Activated microglia fail to affect AMPA receptor-mediated sEPSCs following the blockade of TNFα signaling
Role of Activated Microglia in Neuronal Damage
Inflammatory processes occurring within the CNS trigger a rapid activation of microglia. This activation is evident in situ as such cells show a rapid morphological transition from resting ‘ramified’ cells to amoeboid-activated cells.51 Morphological changes are also accompanied by functional changes as reactive microglia enter in a proliferating phase leading to a substantial increase in their number within the inflamed tissue52 (Figure 1). This activation entails a multitude of metabolic changes sustaining the ability of these cells to function as phagocytes and antigen-presenting cells. In mice with EAE, we detected the first cohort of proliferating microglial cells preceding the onset of the disease,35 reaching a peak of proliferation at 20 days on immunization (d.p.i) and declining during the chronic phase of the disease. We further confirmed using short labeling protocols based on acute 5′Iodio 2′deoxyuridine administration that microglia proliferate actively during the acute but not the chronic phase of EAE (L. Muzio, personal communication). We can only hypothesize, at this stage, that a wave of microglial proliferation is followed by a wave of programmed cell death,53 possibly as a compensatory feedback phenomenon.
Whether or not activation of microglia during the acute phase of EAE is sufficient to induce neuronal cell death is still a matter of debate, as previous studies have convincingly shown that acutely activated microglia produce potentially toxic inflammatory mediators, but chronically activated microglia enhance growth and survival of neural progenitor stem cells and produce immunosuppressive cytokines.54, 55, 56 At this stage, we can only speculate that neurons may undergo programmed cell death at later time points during the course of EAE. Although several studies are in line with this idea,45, 46, 57, 58 the nature of the mechanisms underlying this phenomenon is not well understood. Nevertheless, the morphological and functional changes of microglial cells observed in mice with EAE (Figure 3) clearly support the ensuing idea that activated microglia exert a key role in determining synaptic alterations and possibly neuronal damage. It is also important to note that the relationship between microglial cells and synaptic transmission and damage in MS seems to be particularly complex, as activated microglia in the gray matter, similar to T cells, both regulate and are regulated by glutamate signaling. Activated microglial cells, in fact, are capable of releasing57, 58 and of metabolizing glutamate,57, 58 and in regulating the sensitivity of neuronal AMPA receptors to synaptically released glutamate.57, 58 In addition, stimulation of mGlu 2 receptors (which are upregulated in MS lesions57, 58) enhances microglia-dependent TNFα release and neurotoxicity,16, 17 whereas activation of microglial mGlu 3, 4 and 8 (also upregulated in MS lesions57, 58) has the opposite effect.45 Finally, in white matter MS lesions, microglia/macrophage activity is essential to clear myelin debris, thus promoting remyelination.59

EAE induces microglial activation in the striatum. Striatal microglial activation was consistently activated in mice with EAE (b) compared with healthy controls (a), as shown here by Iba-1 staining at 20 days post-immunization
Role of Inflammatory Cytokines in Synaptic Alteration
Many soluble mediators are released by activated T lymphocytes and microglia during neuroinflammatory diseases, and have the potential to influence synaptic transmission.60, 61, 62, 63 In particular, TNFα, IFNγ and IL-1β are all potential candidates for such an action, as they are released in response to microglial activation,49, 50, 64, 65 increase in the CSF of MS patients,66, 67, 68 and have been shown in vitro to enhance excitatory synaptic transmission60, 62, 69 and downregulate GABA synapses.60 As a matter of fact (i) IL-1β increases glutamate NMDA receptor-mediated synaptic transmission,70, 71 and also regulates AMPA receptor expression and phosphorylation;62 (ii) TNFα increases AMPA receptor expression and synaptic efficacy, an effect also relevant for synaptic plasticity;60, 61 and (iii) IFNγ has been shown to induce excitotoxic damage by enhancing AMPA receptor activity through the formation of a neuron-specific, calcium-permeable receptor complex with the GluR1 subunit of AMPA receptors.69
We have identified TNFα as a critical molecule involved in the synaptic alterations seen in mice with EAE, as we found that TNFα mRNA content increased in EAE brains in parallel to microglial activation.35 Incubation of control brain slices with TNFα mimicked the effects of EAE and of activated microglia on sEPSC kinetic properties, by increasing their decay time and duration. Furthermore, as in the case of the activated microglia, TNFα administration failed to alter other parameters of glutamate transmission, and did not increase further sEPSC duration in slices from mice with acute EAE. Together, these data suggest that TNFα has the potential to promote dendritic spine loss in EAE brains35 through an excitotoxic mechanism. Direct evidence of this process is, however, still lacking.
Finally, to confirm the role of TNFα in the synaptic effects of activated microglia, we performed recordings in slices incubated with both activated primary microglia and TNFR-Ig, blocker of the activity of ambient TNFα. In these experiments, a blockade of TNFα signaling fully prevented the alterations of sEPSCs induced by microglia, suggesting that, at least in vitro, the synaptic effects of microglia are mostly mediated by the TNFα released.35 It is likely, however, that in the in vivo condition, activated microglial cells exert a more complex neuromodulatory action, as they are able to release glutamate itself,72 together with many other secreted molecules (such as IFNγ and IL-1β), which can further modulate the sensitivity of both AMPA and NMDA receptors to their endogenous ligand.49, 50, 64, 65
Role of Inflammatory Cytokines in Neuronal Damage
The inflammatory milieu has often been regarded as tout court hostile to neural cells, and proinflammatory cytokines are considered toxic for neurons and oligodendrocytes. This view has been challenged by newly introduced concepts such as the protective autoimmunity73 and the shift of sense of cytokines during the inflammatory reaction74 underlying the existence of a tissue-protective role for cytokines during recovery from neuroinflammation. Still, at least during the acute phase, where the inflammatory reaction has the main aim of eliminating the alien from the target tissue, the damaging role of cytokines prevails. To this respect, TNFα is paradigmatic. Astrocytes activated by TNFα induce apoptotic oligodendroglial death in vivo75 and, in turn, activate the cerebral endothelium. Microglial cells produce TNFα in the CNS and express as many other CNS cell types including oligodendrocytes, both p75 and p55 receptors.76 It is in fact believed that in these cells TNFα acts as an autocrine-paracrine growth factor. Brain endothelial cells do not produce TNFα but express both the p75 and p55 receptors,77 which, when engaged, determine increased expression of adhesion molecules,78 or determine the release of soluble factors (i.e., prostaglandin E2) active in the brain parenchyma.79 In the white matter, however, TNFα is also capable of exerting beneficial effects in experimental MS, as it promotes remyelination by stimulating the proliferative activity of oligodendrocyte precursor cells through TNFα-receptor-2.80
A neurotoxic role has also been indicated for other prototypical cytokines such as IL-1β, IL6 and IFNγ,81 although it is unclear whether this effect is direct or mediated by other molecules such as reactive oxygen species or glutamate.
Role of Arc/Arg3.1 Gene in CNS-confined Inflammation and Synaptic Changes
The immediate early gene Arc/Arg3.1 is found in the postsynaptic area where it is able to regulate the AMPA receptor trafficking82 and to contribute to the longer phases of LTP.83 Several lines of evidence suggest that its expression is under the control of both the NMDA and AMPA receptors. Indeed, NMDA receptor activation increases its expression levels, whereas AMPAR activation induces its downregulation.84 However, Arc/Arg3.1 expression is also controlled by extracellular signals such as brain-derived neurotrophic factor (BDNF). Indeed, the Arc/Arg3.1 mRNA levels within the granule layer of the dentate gyrus are dramatically upregulated on BDNF brain infusion,85 thus suggesting that diffusible signals may also exert a crucial role in controlling the neuronal function.
As the tissue-specific inflammation associated with EAE is characterized by the expression in situ of a broad number of cytokines, we studied whether the expression levels of Arc/Arg3.1 may be regulated by these molecules. In a first experiment, we evaluated Arc/Arg3.1 mRNA expression levels by using in situ hybridization. In mice with EAE between 20 and 30 d.p.i., Arc/Arg3.1 mRNA levels appeared unchanged within the cerebral cortex and the hippocampus but was dramatically downregulated in the striatum.35 Although the sensitivity of this technique is sufficient to detect small differences in mRNA gene expression, the Arc/Arg3.1 mRNA levels were also evaluated by real-time PCR on striatal microdissections. This analysis confirmed the previous result and showed an approximately 50% reduction in the Arc/Arg3.1 mRNA levels within striata derived from mice affected by EAE.
The inflammatory milieu, which acts within the CNS of mice with EAE, contains a broad number of molecules secreted in part by CNS-infiltrating blood-borne derived cells and in part by activated microglial cells and astrocytes. Thus, we tested whether prototypical Th1 cytokines (IFNγ, TNFα and IL-1β) can modulate the Arc/Arg3.1 mRNA expression on primary neuronal cultures. Within a few hours of administration, these cytokines were capable of significantly reducing the Arc/Arg3.1 mRNA, suggesting a new role for proinflammatory cytokines in controlling the expression of an immediate early gene.
The dramatic downregulation of Arc/Arg3.1 seen in EAE as a consequence of the proinflammatory release by activated microglia prompted us to hypothesize an active role of this early gene in controlling synaptic transmission during experimental MS. Our hypothesis was supported by several lines of evidence. Both the mRNA encoded by the Arc/Arg3.1 gene and the Arc protein accumulate in dendritic spines, where they interact with actin in the control of synaptic transmission.86 The interaction of Arc with dynamin and endophilin, two proteins known to have a role in endocytosis, has also been described.82 Arc and endophilin associate with vesicles that traffic AMPA receptors, and expression of Arc transgene increases the rate of surface AMPA receptor endocytosis and reduces the level of surface AMPA receptors.87 Furthermore, Arc reduces AMPA-mediated sEPSCs, whereas Arc/Arg3.1 overexpression results in a cell-wide decrease in the surface expression of GluR1-containing AMPA receptors.87 By controlling the strength of AMPA-mediated excitatory transmission, Arc expression has an essential role in the induction of homeostatic plasticity,87, 88 a form of long-term synaptic adaptation believed to compensate for Hebbian forms of plasticity (LTP and LTD), by scaling neuronal output without changing the relative strength of individual synapses.89
All these data might indicate that the alterations of sEPSCs seen in mice with EAE during Arc/Arg3.1 downregulation and in response to TNFα reflect the induction of homeostatic plasticity mechanisms, and establishes a strong link between long-lasting synaptic changes and dendritic degeneration in experimental MS.
Conclusions
The effects of soluble mediators, such as cytokines in controlling synaptic activity, have previously been described variably.61 The consequences for human physiology and pathology are however difficult to extrapolate, despite the extreme interest of these findings. As described above, the effects of T cells, microglia and secreted inflammatory cytokines can in fact be both detrimental and neuroprotective, depending on the duration of the inflammatory process and on its preferential location in the white or gray matter. The case of BDNF appears to be particularly significant in this respect. BDNF is in fact released by activated T cells, microglia and astrocytes in MS lesions, where it promotes remyelination and neuronal and oligodendroglial survival.90, 91, 92 On the other hand, increased BDNF release in the gray matter by immune cells and by neurons might favor excitotoxic neuronal damage during the acute phase of neuroinflammatory diseases, as BDNF increases glutamate release93, 94 and controls AMPA and NMDA receptor phosphorylation, expression and trafficking.95, 96 It also favors LTP of excitatory transmission, whereas it preferentially inhibits GABA release.95, 96, 97 Together, these findings indicate that BDNF exerts complex and opposite effects in the white and gray matter of patients with MS. According to this conclusion, increased BDNF release has been claimed to mediate the therapeutic effects of MS-specific medications on tissue destruction in white matter inflammatory lesions,59 whereas MS subjects with impaired BDNF signaling exhibit less severe gray matter atrophy.98
The investigation of the cross-talk between the immune system and neurons using synaptic activity, glutamate excitotoxity, synapse and spine integrity, and neuron-glia interaction as primary readouts has to be taken as a step forward, and put in the context of experimental and human pathology. The results that we have presented in this review article are mainly obtained in a mouse model of MS, but they already have potential clinical consequences, in terms of comprehension of pathogenic mechanisms and in the possible identification of novel therapeutic targets for MS. The synaptopathy that we and others described to be concomitant to, and caused by, neuroinflamation may constitute, when prolonged enough and pushed beyond a possible point of no return, the long searched for link between inflammation and neurodegeneration in chronic diseases of the CNS such as MS. It may also help to explain, along with many other pathological events, the early cognitive deficits seen frequently in some MS patients, and may explain the correlation that has been described5 between the amount of inflammation in the early phases of the disease and the rate of progression in the late, so-called neurodegenerative, phase.
We can further speculate that synaptic changes occurring early during immune-mediated inflammatory demyelinating disorders of the CNS – caused by pro-inflammatory molecules released by activated blood-borne or CNS-resident immune cells – might also contribute to limiting plastic compensatory ‘protective’ changes (e.g., driven by developmental molecules or stem/precursor cells) occurring within neural networks. This can be considered as a contributing phenomenon to foster the transition of CNS-confined inflammatory neurological disorders into chronic irreversible and progressive neurodegenerative disorders.
We firmly believe that new investigations in the forthcoming years will shed further light on the link between inflammation, synaptic transmission and neuronal damage, and that soon these results will be taken to the bedside of patients affected by neuroinflammatory disorders.
Abbreviations
- BDNF:
-
brain-derived neurotrophic factor
- CNS:
-
central nervous system
- EAE:
-
experimental autoimmune encephalomyelitis
- EPSC:
-
excitatory postsynaptic current
- LTD:
-
long-term depression
- LTP:
-
long-term potentiation
- mGlu receptor:
-
metabotropic glutamate receptor
- MS:
-
multiple sclerosis
- NO:
-
nitric oxide
References
Zipp F, Aktas O . The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci 2006; 29: 518–527.
Centonze D, Finazzi-Agrò A, Bernardi G, Maccarrone M . The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci 2007; 28: 180–187.
Compston A, Coles A . Multiple sclerosis. Lancet 2008; 372: 1502–1517.
Trapp BD, Nave KA . Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 2008; 31: 247–269.
Geurts JJ, Barkhof F . Grey matter pathology in multiple sclerosis. Lancet Neurol 2008; 7: 841–851.
Allan SM, Tyrrell PJ, Rothwell NJ . Interleukin-1 and neuronal injury. Nat Rev Immunol 2005; 5: 629–640.
Linker RA, Rott E, Hofstetter HH, Hanke T, Toyka KV, Gold R . EAE in beta-2 microglobulin-deficient mice: axonal damage is not dependent on MHC-I restricted immune responses. Neurobiol Dis 2005; 19: 218–228.
Babcock AA, Kuziel WA, Rivest S, Owens T . Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 2003; 23: 7922–7930.
Rocca MA, Absinta M, Valsasina P, Ciccarelli O, Marino S, Rovira A et al. Abnormal connectivity of the sensorimotor network in patients with MS: a multicenter fMRI study. Hum Brain Mapp 2009; 30: 2412–2425.
Hauser SL, Oksenberg JR . The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron 2006; 52: 61–76.
Waxman SG . Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 2006; 7: 932–941.
Forder JP, Tymianski M . Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience 2009; 158: 293–300.
Linden DJ, Dickinson MH, Smeyne M, Connor JA . A long-term depression of AMPA currents in cultured cerebellar Purkinje neurons. Neuron 1991; 7: 81–89.
Gubellini P, Saulle E, Centonze D, Bonsi P, Pisani A, Bernardi G et al. Selective involvement of mGlu1 receptors in corticostriatal LTD. Neuropharmacology 2001; 40: 839–846.
Calabresi P, Centonze D, Pisani A, Bernardi G . Metabotropic glutamate receptors and cell-type-specific vulnerability in the striatum: implication for ischemia and Huntington's disease. Exp Neurol 1999; 158: 97–108.
Taylor DL, Jones F, Kubota ES, Pocock JM . Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci 2005; 25: 2952–2964.
Pinteaux-Jones F, Sevastou IG, Fry VA, Heales S, Baker D, Pocock JM . Myelin-induced microglial neurotoxicity can be controlled by microglial metabotropic glutamate receptors. J Neurochem 2008; 106: 442–454.
Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D . Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3T. Brain 2005; 128 (Part 5): 1016–1025.
Cianfoni A, Niku S, Imbesi SG . Metabolite findings in tumefactive demyelinating lesions utilizing short echo time proton magnetic resonance spectroscopy. AJNR Am J Neuroradiol 2007; 28: 272–277.
Stover JF, Lowitzsch K, Kempski OS . Cerebrospinal fluid hypoxanthine, xanthine and uric acid levels may reflect glutamate-mediated excitotoxicity in different neurological diseases. Neurosci Lett 1997; 238: 25–28.
Sarchielli P, Greco L, Floridi A, Floridi A, Gallai V . Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Neurol 2003; 60: 1082–1088.
Newcombe J, Uddin A, Dove R, Patel B, Turski L, Nishizawa Y et al. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol 2008; 18: 52–61.
Geurts JJ, Wolswijk G, Bö L, van der Valk P, Polman CH, Troost D et al. Altered expression patterns of group I and II metabotropic glutamate receptors in multiple sclerosis. Brain 2003; 126 (Pt 8): 1755–1766.
Geurts JJ, Wolswijk G, Bö L, Redeker S, Ramkema M, Troost D et al. Expression patterns of Group III metabotropic glutamate receptors mGluR4 and mGluR8 in multiple sclerosis lesions. J Neuroimmunol 2005; 158: 182–190.
Vallejo-Illarramendi A, Domercq M, Pérez-Cerdá F, Ravid R, Matute C . Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis 2006; 21: 154–164.
Hardin-Pouzet H, Krakowski M, Bourbonnière L, Didier-Bazes M, Tran E, Owens T . Glutamate metabolism is down-regulated in astrocytes during experimental allergic encephalomyelitis. Glia 1997; 20: 79–85.
Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y et al. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol 2002; 125: 170–178.
Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL et al. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 2007; 66: 732–739.
Matute C, Alberdi E, Domercq M, Pérez-Cerdá F, Pérez-Samartín A, Sánchez-Gómez MV . The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci 2001; 24: 224–230.
Pitt D, Werner P, Raine CS . Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000; 6: 67–70.
Smith T, Groom A, Zhu B, Turski L . Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 2000; 6: 62–66.
Wallström E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T . Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci 1996; 137: 89–96.
Bolton C, Paul C . MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther 1997; 282: 397–402.
Plaut GS . Effectiveness of amantadine in reducing relapses in multiple sclerosis. J R Soc Med 1987; 80: 91–93.
Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci 2009; 29: 3442–3452.
Gilgun-Sherki Y, Panet H, Melamed E, Offen D . Riluzole suppresses experimental autoimmune encephalomyelitis: implications for the treatment of multiple sclerosis. Brain Res 2003; 989: 196–204.
Caramia MD, Palmieri MG, Desiato MT, Boffa L, Galizia P, Rossini PM et al. Brain excitability changes in the relapsing and remitting phases of multiple sclerosis: a study with transcranial magnetic stimulation. Clin Neurophysiol 2004; 115: 956–965.
Piani D, Frei K, Do KQ, Cuénod M, Fontana A . Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 1991; 133: 159–162.
Werner P, Pitt D, Raine CS . Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 2001; 50: 169–180.
Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF . Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 2000; 20: 89–102.
Centonze D, Bari M, Rossi S, Prosperetti C, Furlan R, Fezza F et al. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain 2007; 130 (Part 10): 2543–2553.
Rossi S, Furlan R, De Chiara V, Musella A, Lo Giudice T, Mataluni G et al. Exercise attenuates the clinical, synaptic and dendritic abnormalities of experimental autoimmune encephalomyelitis. Neurobiol Dis 2009; 36: 51–59.
Fazio F, Notartomaso S, Aronica E, Storto M, Battaglia G, Vieira E et al. Switch in the expression of mGlu1 and mGlu5 metabotropic glutamate receptors in the cerebellum of mice developing experimental autoimmune encephalomyelitis and in autoptic cerebellar samples from patients with multiple sclerosis. Neuropharmacology 2008; 55: 491–499.
Lewitus GM, Zhu J, Xiong H, Hallworth R, Kipnis J . CD4(+)CD25(−) effector T cells inhibit hippocampal long-term potentiation in vitro. Eur J Neurosci 2007; 26: 1399–1406.
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009; 132 (Part 5): 1175–1189.
Schori H, Yoles E, Schwartz M . T-cell-based immunity counteracts the potential toxicity of glutamate in the central nervous system. J Neuroimmunol 2001; 119: 199–204.
Lombardi G, Miglio G, Canonico PL, Naldi P, Comi C, Monaco F . Abnormal response to glutamate of T lymphocytes from multiple sclerosis patients. Neurosci Lett 2003; 340: 5–8.
Maragakis NJ, Rothstein JD . Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2006; 2: 679–689.
Schwartz M, Butovsky O, Brück W, Hanisch UK . Microglial phenotype: is the commitment reversible? Trends Neurosci 2006; 29: 68–74.
Muzio L, Martino G, Furlan R . Multifaceted aspects of inflammation in multiple sclerosis: The role of microglia. J Neuroimmunol 2007; 191: 39–44.
Streit WJ, Kreutzberg GW . Lectin binding by resting and reactive microglia. J Neurocytol 1987; 16: 249–260.
Ponomarev ED, Shriver LP, Maresz K, Dittel BN . Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res 2005; 81: 374–389.
Gehrmann J, Banati RB . Microglial turnover in the injured CNS: activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J Neuropathol Exp Neurol 1995; 54: 680–688.
Aarum J, Sandberg K, Haeberlein SL, Persson MA . Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci USA 2003; 100: 15983–15988.
Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N et al. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest 2006; 116: 905–915.
Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, Marshall IInd GP et al. Microglia instruct subventricular zone neurogenesis. Glia 2006; 54: 815–825.
Polazzi E, Contestabile A . Overactivation of LPS-stimulated microglial cells by co-cultured neurons or neuron-conditioned medium. J Neuroimmunol 2006; 172: 104–111.
Neumann H . Control of glial immune function by neurons. Glia 2001; 36: 191–199.
Graber JJ, Dhib-Jalbut S . Protective autoimmunity in the nervous system. Pharmacol Ther 2009; 121: 147–159.
Stellwagen D, Beattie EC, Seo JY, Malenka RC . Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 2005; 25: 3219–3228.
Stellwagen D, Malenka RC . Synaptic scaling mediated by glial TNF-alpha. Nature 2006; 440: 1054–1059.
Lai AY, Swayze RD, El-Husseini A, Song C . Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol 2006; 175: 97–106.
Cumiskey D, Curran BP, Herron CE, O’Connor JJ . A role for inflammatory mediators in the IL-18 mediated attenuation of LTP in the rat dentate gyrus. Neuropharmacology 2007; 52: 1616–1623.
Block ML, Hong JS . Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 2005; 76: 77–98.
Kawanokuchi J, Mizuno T, Takeuchi H, Kato H, Wang J, Mitsuma N et al. Production of interferon-gamma by microglia. Mult Scler 2006; 12: 558–564.
Rovaris M, Barnes D, Woodrofe N, du Boulay GH, Thorpe JW, Thompson AJ et al. Patterns of disease activity in multiple sclerosis patients: a study with quantitative gadolinium-enhanced brain MRI and cytokine measurement in different clinical subgroups. J Neurol 1996; 243: 536–542.
Khademi M, Wallström E, Andersson M, Piehl F, Di Marco R, Olsson T . Reduction of both pro- and anti-inflammatory cytokines after 6 months of interferon beta-1a treatment of multiple sclerosis. J Neuroimmunol 2000; 103: 202–210.
Baraczka K, Pozsonyi T, Szüts I, Ormos G, Nékám K . Increased levels of tumor necrosis alpha and soluble vascular endothelial adhesion molecule-1 in the cerebrospinal fluid of patients with connective tissue diseases and multiple sclerosis. Acta Microbiol Immunol Hung 2003; 50: 339–348.
Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J 2008; 22: 1797–1806.
Yang J, Woodhall GL, Jones RS . Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci 2006; 26: 406–410.
Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 2003; 23: 8692–8700.
Cannella B, Raine CS . The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol 1995; 37: 424–435.
Schwartz M, London A, Shechter R . Boosting T-cell immunity as a therapeutic approach for neurodegenerative conditions: the role of innate immunity. Neuroscience 2009; 158: 1133–1142.
Martino G, Adorini L, Rieckmann P, Hillert J, Kallmann B, Comi G et al. Inflammation in multiple sclerosis: the good, the bad, and the complex. Lancet Neurol 2002; 1: 499–509.
Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G et al. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol 1998; 153: 801–813.
Sawada M, Kondo N, Suzumura A, Marunouchi T . Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res 1989; 491: 394–397.
Osburg B, Peiser C, Dömling D, Schomburg L, Ko YT, Voigt K et al. Effect of endotoxin on expression of TNF receptors and transport of TNF-alpha at the blood-brain barrier of the rat. Am J Physiol Endocrinol Metab 2002; 283: E899–E908.
Omari KM, Dorovini-Zis K . CD40 expressed by human brain endothelial cells regulates CD4+ T cell adhesion to endothelium. J Neuroimmunol 2003; 134: 166–178.
Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A . Inflammatory response: pathway across the blood-brain barrier. Nature 2001; 410: 430–431.
Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP . TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001; 4: 1116–1122.
Bezzi P, Volterra A . A neuron-glia signalling network in the active brain. Curr Opin Neurobiol 2001; 11: 387–394.
Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 2006; 52: 445–459.
Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF et al. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci 2000; 20: 3993–4001.
Rao VR, Pintchovski SA, Chin J, Peebles CL, Mitra S, Finkbeiner S . AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci 2006; 9: 887–895.
Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV et al. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 2002; 22: 1532–1540.
Bramham CR, Worley PF, Moore MJ, Guzowski JF . The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci 2008; 28: 11760–11767.
Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006; 52: 475–484.
Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT . Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron 2006; 52: 461–474.
Turrigiano G . Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol 2007; 17: 318–324.
Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE et al. Activated human T cells B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 1999; 189: 865–870.
Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H . The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. J Neuroimmunol 2000; 107: 161–166.
Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H . BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 2002; 125 (Part 1): 75–85.
Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM . Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA 1998; 95: 10884–10889.
Numakawa T, Matsumoto T, Adachi N, Yokomaku D, Kojima M, Takei N . Brain-derived neurotrophic factor triggers a rapid glutamate release through increase of intracellular Ca(2+) and Na(+) in cultured cerebellar neurons. J Neurosci Res 2001; 66: 96–108.
Carvalho AL, Caldeira MV, Santos SD, Duarte CB . Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br J Pharmacol 2008; 153 (Suppl 1): S310–S324.
Waterhouse EG, Xu B . New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci 2009; 42: 81–89.
Frerking M, Malenka RC, Nicoll RA . Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol 1998; 80: 3383–3386.
Zivadinov R, Weinstock-Guttman B, Benedict R, Tamaño-Blanco M, Hussein S, Abdelrahman N et al. Preservation of gray matter volume in multiple sclerosis patients with the Met allele of the rs6265 (Val66Met) SNP of brain-derived neurotrophic factor. Hum Mol Genet 2007; 16: 2659–2668.
Acknowledgements
This investigation was supported by the Italian National Ministero dell’Università e della Ricerca to DC, by the Italian National Ministero della Salute to DC, by Fondazione Italiana Sclerosi Multipla (FISM) to DC, RF, LM and GM, and by BMW to GM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Rights and permissions
About this article
Cite this article
Centonze, D., Muzio, L., Rossi, S. et al. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ 17, 1083–1091 (2010). https://doi.org/10.1038/cdd.2009.179
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.179
Keywords
This article is cited by
-
Glia Connect Inflammation and Neurodegeneration in Multiple Sclerosis
Neuroscience Bulletin (2023)
-
Cladribine treatment improves cortical network functionality in a mouse model of autoimmune encephalomyelitis
Journal of Neuroinflammation (2022)
-
The impact of white matter hyperintensities on speech perception
Neurological Sciences (2020)
-
Cytokine inflammatory threat, but not LPS one, shortens GABAergic synaptic currents in the mouse spinal cord organotypic cultures
Journal of Neuroinflammation (2019)
-
Bridging pro-inflammatory signals, synaptic transmission and protection in spinal explants in vitro
Molecular Brain (2018)
