
Abstract
Bcl-2 family proteins regulate mitochondrial apoptosis downstream of diverse stressors. Cancer cells frequently deregulate Bcl-2 proteins leading to chemoresistance. We have optimized a platform for solid tumors in which Bcl-2 family resistance patterns are inferred. Functional mitochondria were isolated from neuroblastoma (NB) cell lines, exposed to distinct BH3-domain peptides, and assayed for cytochrome c release. Such BH3 profiles revealed three patterns of cytochrome c response. A subset had a dominant NoxaBH3 response implying Mcl1 dependence. These cells were more sensitive to small molecules that antagonize Mcl1 (AT-101) than those that antagonize Bcl-2, Bcl-xL and Bcl-w (ABT-737). A second subset had a dominant BikBH3 response, implying a Bcl-xL/-w dependence, and was exquisitely sensitive to ABT-737 (IC50 <200 nM). Finally, most NB cell lines derived at relapse were relatively resistant to pro-death BH3 peptides and Bcl-2 antagonists. Our findings define heterogeneity for apoptosis resistance in NB, help triage emerging Bcl-2 antagonists for clinical use, and provide a platform for studies to characterize post-therapy resistance mechanisms for NB and other solid tumors.
Similar content being viewed by others

Main
The Bcl-2 family of proteins governs mitochondrial apoptosis. Specific cellular stressors, including those initiated by chemo- and radiotherapy, activate select pro-death BH3-only proteins through diverse transcriptional or translational mechanisms.1 After activation, these proteins are free to interact with multi-domain Bcl-2 family members residing at the outer mitochondrial membrane. Here, a subset of BH3-only proteins can directly activate the obligate executioners Bak or Bax, inducing oligomerization and cytochrome c release, committing the cell to death. BH3-only proteins with this capacity have been termed ‘activator’ BH3s and include Bid and Bim. Alternatively, they may be sequestered within the hydrophobic pocket of pro-survival proteins such as Mcl1, Bcl-2, Bcl-xL, Bcl-w, and A1/Bfl, neutralizing their death signal. Other BH3-only proteins (such as Noxa, Bik, Bad, and others) appear incapable of direct Bak/Bax activation but enable apoptosis indirectly. This ‘enabler’ function has been variably attributed to a higher affinity for pro-survival pockets causing displacement of sequestered activator BH3s or to displacement of pro-survival Bcl-2 proteins from direct Bak/Bax repression. In either case the stoichiometry and functional state of Bcl-2 family members determine whether an apoptotic signal is transduced.
Distinct tissues heterogeneously express Bcl-2 proteins, allowing tissue-specific responses to encountered stressors. Aberrant Bcl-2 family expression may also accompany tumor progression further contributing to tumor-specific variability in these pathways. For example, hematopoeitic malignancies frequently overexpress a single pro-survival Bcl-2 member and are exquisitely sensitive to its selective antagonism.2, 3, 4, 5 Indeed, these malignancies are often responsive to multiple retrieval therapies after relapse. In contrast, solid tumors are generally more apoptosis resistant, commonly failing front line and/or salvage therapy because of their ability to attenuate apoptotic signaling.6 Therefore, further investigation into the mechanisms of apoptosis evasion for solid tumors is warranted.
Synthetic peptides consisting of BH3 domains from BH3-only proteins can be used as bioprobes to assess mitochondrial responses to death stimuli.7, 8, 9, 10 By profiling isolated cancer cell mitochondria for cytochrome c release after exposure to a diverse panel of BH3 peptides that have distinct affinities for the Bcl-2 pro-survival proteins, Certo et al.7 identified pro-survival addiction patterns in a model of leukemia that was distinct from normal hematopoeitic tissues. BH3 profiling was used to define resistance mechanisms in these cancers and revealed Bcl-2 dependence for ALL, CLL, and certain lymphomas.2, 3, 4 BH3 profiling has neither been applied to solid tumor investigations, nor any pediatric tumor to date. Neuroblastoma (NB) is a highly lethal pediatric solid tumor derived from developing sympathetic neuroblasts. NBs require intact mitochondrial apoptosis for chemotherapy-induced cell death to occur.11, 12 Evasion of apoptosis contributes to its aggressive phenotype13 and patients frequently succumb to chemoresistant disease.11 We therefore sought to define the major patterns of apoptosis response and resistance in NB using an unbiased functional mitochondria-based assay.
We optimized a BH3 profiling approach for solid tumors and show that sufficient functional mitochondria can be collected from adherent cell lines or freshly obtained xenografts. NB mitochondria respond to diverse BH3 domains with cytochrome c release in highly reproducible patterns (called a BH3 response profile). These patterns are distinct from non-malignant cells that do not respond to enabler BH3 peptides, supporting a primed-for-death state in NB. Further, evidence for Bcl-2 pro-survival protein redundancy and heterogeneity within this single cancer type is shown. At least three distinct patterns were identified, allowing the predominant pro-survival dependence to be inferred. Finally, mitochondrial BH3 response profiles were highly correlated with, and predictive of, whole cell responses to small molecule Bcl-2 family antagonists. Our data suggest that BH3 profiles reliably capture the Bcl-2 family-governed apoptotic set point for these tumors. These studies elucidate key pathways of apoptosis evasion in NB and may be used to define surrogate biomarkers to triage small molecule Bcl-2 antagonists. They also provide a platform for mechanistic studies to characterize therapy resistance in this tumor type.
Results
BH3 response profiles reveal distinct pro-survival addiction patterns in neuroblastoma
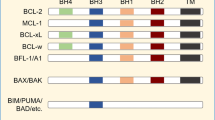
We isolated mitochondria from 10 different tumor-derived NB cell lines representative of high-risk disease. Mitochondria were exposed to saturating concentrations of BH3-domain peptides and assessed for cytochrome c release as a marker of mitochondrial outer membrane permeability (MOMP) and apoptosis. Test peptides encompassed the α-helical death domains and, based on previous studies, maintain selective affinity for pro-survival BH pockets. Therefore, the BH3 response profile defines the tumor cell set point for transducing BH3-mediated death stimuli. For example, cytochrome c release in response to NoxaBH3 suggests an Mcl1 survival dependence, as Mcl1 activity alone is neutralized by NoxaBH3. Table 1 summarizes previously determined BH3 peptide/pro-survival protein affinities. Such affinities are largely concordant across diverse methodologies.7, 14
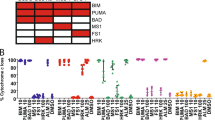
All NB-derived mitochondria released cytochrome c in response to recombinant tBid (positive control), BidBH3, and BimBH3 peptides (Figure 1). Cytochrome c release in response to such ‘activator’ peptides confirms intact Bak/Bax signal transduction, and with few exceptions (see SK-N-AS) this release was robust. In contrast, the substituted BidaltBH3 peptide was inactive against all cell line mitochondria. BH3 peptide responses were reproducible in replicate experiments as rank order potency for the peptides was maintained (with the exception of BmfBH3), reflecting a specific functional readout. Cytochrome c release to at least a subset of enabler peptides (e.g. Bik, Noxa, Bmf) was found in the majority of NBs tested, suggesting these mitochondria harbor endogenous activator BH3 signals that are tonically suppressed. Furthermore, no enabler BH3 peptides elicited cytochrome c release from non-transformed neural RPE1 cells or human T cells (Figure 1c).

BH3 response profiling defines three distinct pro-survival addiction patterns in NB. Functional mitochondria were exposed to a saturating concentration of individual BH3-only death domain peptides and analyzed by ELISA immunoassay for cytochrome c release. Gray bars, putative activator BH3 peptides; black bars, enabler BH3 peptides with selective binding to pro-survival proteins; see Table 1. BH3 response profiles showed a predominant Noxa-induced cytochrome c release group (a), a dominant Bik-induced cytochrome c release group (b), and tumor cells relatively resistant to enabler peptides (c). The latter group is enriched for cell lines established at relapse. Control RPE-1 and T-cell mitochondrial responses are also shown. Average and standard deviations for at least two biological and technical replicates are shown. Rank order responsiveness to peptides was identical between independent experiments. hTcells, human T-lymphocytes; μM, micromolar; BH3-only protein name represents the BH3-domain peptide from that protein; Tbid, recombinant truncated Bid protein; Bidalt, substituted BidBH3 domain to abrogate Bcl-2-family interactions
As BH3 peptides have redundant binding affinities for Bcl-2 members, we chose to assess the entire repertoire of BH3 responses as indicative of the mitochondrial signaling set point. We therefore performed hierarchical clustering to characterize these response patterns. Clustering was initially performed both with all peptide responses included (n=9 peptides) as well as omitting Bmf (n=8 peptides). We reasoned that the physiochemical stress of adherent cell collection might lead to activation of endogenous Bmf, a key arbiter of anoikis.15 We also noted that Bmf-mediated cytochrome c release was much more variable between biological replicates compared with other peptides. Clustering of BH3 response profiles from NB mitochondria identified three predominant clades, or BH3 response classes (Figure 2a). Both clustering analyses (including or omitting Bmf) were near identical with only one cell line (LAN5) shifting clusters in this comparison. Moreover, assessing the dendrograms revealed that most of the informativeness in clustering was derived from the differential responses to NoxaBH3 and BikBH3, as reclustering using only these two peptides recapitulated the exact same clade inclusion as the 8 peptide analysis (Figure 2a; data not shown).

(a) Hierarchical clustering analysis of NB cell lines and xenografts based on BH3 profiles reveals three distinct response patterns. Shaded clades delineate the three clusters of response to the two most dominant enabler peptides, Noxa and Bik. ‘SK-N-AS- (POST VP-16)’ represents the BH3 profile of SK-N-AS after sublethal etoposide treatment. ‘XG’, xenograft, profiles show all but one NB xenograft clustered with its parent monolayer cell line response. (b) Xenograft and monolayer culture BH3 profiles show concordance. Non-necrotic xenograft tumors <500 mm3 were collected from the flanks of immunodeficient mice. Tumors were bisected with one section disassociated and plated to establish a monolayer cell line for mitochondrial BH3 response profiling (monolayer; left panels). The other section was immediately used to isolate the heavy membrane fraction containing functional mitochondrial for xenograft BH3 response profiling (right panels). Error bars represent the average and standard deviations of at least two separate experiments
NBs cells from the same cluster showed similar BH3 response patterns (Figures 1 and 2). The most potent enabler peptides for each class could be visually determined and statistically validated. We compared cytochrome c release mediated by each enabler BH3 peptide with release mediated by the activator BH3 peptides (tBid, Bim, or Bid) for each cell line. Enabler peptides with cytochrome c release that exceeded the negative control (BidAltBH3) and that were statistically indistinguishable from responses to activator BH3s or tBid were considered robust releasers (e.g. NoxaBH3 in IMR5; Figure 1a). Peptides with cytochrome c release intermediate between the positive control and negative control were considered active but less potent (e.g. BikBH3 in IMR5); and those statistically indistinguishable from the negative control were non-active. This allowed us to infer pro-survival Bcl-2 protein dependence patterns among these classes.
The first cluster (that includes IMR5, NLF, and 902R) showed the most robust cytochrome c release with NoxaBH3 (called Noxa-dominant), supporting an Mcl1 survival dependence (Figure 1a). Noxa-induced cytochrome c release for these cells was indistinguishable from direct activator peptides tBid, BidBH3, and BimBH3 (P>0.20 for each), indicative of robust release. These cells typically also had a demonstrable response to BikBH3, suggesting a potential co-dependence on either Bcl-xL or Bcl-w, though this response was less robust (reduced compared with positive control release; P<0.05). A dependence on Bcl-2 is less likely as BadBH3 did not induce significant cytochrome c release. However, BikBH3 also has modest Mcl1 affinity, which may explain its effect at the high concentrations studied (Table 1). Overall, the profile is supportive of a dominant Mcl1 addiction pattern and predicts therapies that antagonize Mcl1 may augment cell death in these cells.
The second cluster included LAN5, SMS-SAN, SMS-KCN, and NB1643. Mitochondria from these cells were most sensitive to BikBH3 (called Bik-dominant) (Figures 1b and 2). NoxaBH3 responses were present in these cell lines but less robust (P<0.01 compared with positive controls). Except for LAN5, BmfBH3 also triggered cytochrome c release consistent with its similar Bcl-2 family affinities (Table 1). All but SMS-SAN were unresponsive to BadBH3, suggesting Bcl-2 is not the principal survival mediator, but suggesting a Bcl-xL or Bcl-w dependence. These cells are predicted to be sensitive to Bcl-2/Bcl-xL/Bcl-w antagonists such as ABT-737.
The third cluster was characterized by relative resistance to all enabler BH3 peptides (represented by CHP134, SK-N-AS, and BE2C; see Figures 1c and 2). In the hierarchical clustering, the greatest distinction in response profiles was between this subset of enabler resistant cells and the other two groups (Figure 2a). SK-N-AS cells also had consistently attenuated responses to activator BH3 peptides and may have resistance mechanisms in place downstream of Bcl-2 signaling. All resistant cell lines expressed Bak and Bax protein (Figure 4) even at the mitochondria (from isolated mitochondria immunoblots; data not shown), suggesting that they are not primed for apoptosis. Notably, all such BH3-resistant NB cell lines were derived from tumors obtained at relapse after cytotoxic therapies. This is consistent with the extreme therapy resistance seen clinically after relapse after high-risk therapy, which is uniformly lethal.

Protein expression patterns of multi-domain Bcl-2 family proteins in NB. (a) NB cells growing in replete media at steady state were collected and immunoblotted for proteins. β-tubulin served as loading control. *Cells with a Bik-dominant BH3 profile. (b) Histograms representing Bcl-2 family protein immunoblots normalized to β-tubulin loading control
Neural RPE1 cells and T cells are non-transformed and show a lack of enabler BH3 responses (Figure 1c). Bax/Bak signaling was intact, as their mitochondria released cytochrome c in response to BimBH3 (though to a lesser degree than cancer-derived mitochondria) potentially reflecting differences in Bax and/or Bak pre-activation. Overall, the resistance of such normal cells to enabler BH3 peptides reinforces the notion that NB cells are more dependent on apoptosis suppression, providing a potential therapeutic window for agents that antagonize pro-survival Bcl-2 family functions.
In vivo tumor formation does not alter BH3 response profiles
It is possible that artifactual priming with endogenous activator BH3 proteins occurs with cell handling and does not reflect the inherent apoptotic signaling status of the cancer. That non-transformed cells did not respond to enabler BH3 peptides was reassuring, particularly as RPE1 cells are adherent and also underwent mechanical and chemical (versene) cytoskeletal stress during collection. We sought to determine whether cell/cell cues that exist in three-dimensional tumor structures, and include heterotypic cells, might alter response profiles. Survival signals present in the microenvironment might prevent cytochrome c release by repressing endogenous BH3 protein activation. We obtained BH3 response profiles from mitochondria collected directly from fresh xenografts for three NB cell lines. Xenograft mitochondrial responses did not differ qualitatively from those obtained under monolayer conditions (Figure 2b). Although xenograft cytochrome c release was marginally blunted in comparison with monolayer results, the rank order responsiveness across peptides was identical in all three cell lines. Hierarchical clustering placed both SK-N-AS and SMS-SAN xenografts in the same class as their monolayer responses, whereas NB-1643 was placed in the BH3-resistant class (Figure 2a). That xenografts retained relatively robust enabler BH3 peptide responses (see SMS-SAN) lessens the likelihood that its primed status in vitro arises solely because of substratum detachment. These studies importantly show the feasibility of obtaining BH3 response profiles from freshly isolated tumor material as well.
BH3 peptide responses act through genetically defined roles of BH3 proteins in the intrinsic apoptosis pathway
BH3 peptide concentrations used for cytochrome c release assays are sufficient to saturate Bcl-2 protein-binding sites and promote competitive displacement of activator BH3 proteins. To test this we exposed mitochondria to decreasing concentrations of both enabler and activator BH3 peptides, Bik, Noxa, and Bim (see LAN5, Figure 3a). These results support saturation as higher concentrations result in a plateau in cytochrome c release. These findings are also consistent with the distinction between activator and enabler BH3 effects. Minimal Bim exposures induced cytochrome c release (1 μM), presumably through direct interaction with Bax/Bak, and 5 μM was fully active (compared with tBid). In contrast, higher Bik and Noxa peptide concentrations were required to induce a similar degree of cytochrome c release and the effect plateaued at 25 μM. Further, Bcl-xL/-w-dependent cell lines (e.g. LAN5, Bik-dominant) responded to Bik at 1 μM, and robustly at 5 μM, whereas higher Noxa concentrations were required. This is consistent with differing potency because of selective pro-survival protein affinities. As our findings suggest that priming for cell death has occurred in many NB cells at steady state, we sought evidence for tonic repression of activator BH3 proteins. Co-immunoprecipitation experiments demonstrated Bim, but not Bad, Puma, tBid, or Bak, to be bound to Mcl1, Bcl-xL, and Bcl-2, further supporting the theory that they harbor endogenous activator BH3 proteins sequestered to pro-survival members to prevent apoptosis (Figure 3b).

Functional validation of BH3 peptide activity. (a) Dose-dependent response of LAN5 mitochondria to BH3 peptides BimBH3, BikBH3, and NoxaBH3 showing enhanced sensitivity to activator (Bim) peptides, and showing higher potency for Bik over Noxa in this Bik-dominant NB. (b) Co-immunoprecipitation data confirm that NBs are primed for death as endogenous activated BH3 protein Bim is bound to Mcl1, Bcl-2, and Bcl-xL at steady state. Reverse pull down, β-tubulin IP, and beads lane confirm these to be true BH protein interactions. Bim-EL, ‘Extra long’ Bim isoform; Bim-L, ‘Long’ isoform. (c) SK-N-AS shows change in BH3 profile to a Noxa-dominant pattern after exposure to sublethal doses of etoposide, identifying Mcl1 as possible contributer to etoposide resistance in this cell line; 2 × 108 cells were exposed to 10 μg/ml etoposide, then cells were washed and collected, mitochondria isolated, and immediately exposed to the panel of BH3 peptides to evaluate cytochrome c release
We next sought to determine whether a cell line absent demonstrable priming for death could become primed after cytotoxic drug exposure. Etoposide has been shown to activate Bim to induce apoptosis.16 SK-N-AS cells, established at relapse after chemotherapy, are resistant to etoposide (IC50 by MTT >150 μg/ml). They cluster with other ‘resistant’ NB cell lines based on BH3 response profiles (Figure 3c). We pretreated SK-N-AS cells with a sublethal etoposide exposure (10 μg/ml) and assessed BH3 responses (Figure 3c). Earlier ineffective peptides, Noxa and Bik, more potently induced cytochrome c release after exposure to etoposide at >1 log less than the IC50. This suggests that pro-survival Bcl-2 proteins such as Mcl1 become primed with endogenous activator BH3-only protein downstream of this stress stimulus. Consistent with this, repeated hierarchical cluster analysis assigned SK-N-AS after etoposide treatment to the Noxa-dominant group (Figure 2a).
Pro-survival Bcl-2 family protein addiction patterns are not evident in whole cell immunoblots
Immunoblots of multi-domain Bcl-2 proteins from NB whole cell lysates demonstrated variable and redundant pro-survival protein expression (Figure 4). Most expressed Bax and Bak at readily detectable levels, whereas KCN and SMS-SAN had reduced Bak expression. Of these, SMS-SAN cells showed robust cytochrome c release in our functional assays suggesting that Bak is not required for this process (e.g. Bax may suffice) or that the amount of Bak present is sufficient. For pro-survival Bcl-2 proteins, expression levels did not seem to define mitochondrial resistance to BH3 peptides, as SK-N-AS, CHP134, and BE2C did not markedly overexpress any particular pro-survival protein relative to sensitive cell lines. However, there was a notable reduction in Mcl1 expression defining cells with BikBH3-sensitive mitochondria (Bik-dominant). Bak protein levels are also reduced in these same cells possibly through loss of the stabilization influence of Mcl1 on Bak.17 Therefore, low Mcl1 protein may be a biomarker of a Bik-dominant response profile.
BH3 profiling predicts sensitivity to Bcl-2 family antagonists
To validate that BH3 profiling accurately identified cellular Bcl-2 family pro-survival dependence, we treated NB cells in vitro with small molecule antagonists of Bcl-2 proteins. ABT-737 (Abbott Laboratories, Abbott Park, IL, USA) binds to Bcl-2, Bcl-xL, and Bcl-w with subnanomolar affinity (Ki<1 nM), whereas AT-101 (Ascenta Therapeutics, Malvern, PA, USA) binds to Bcl-2 and Bcl-xL less potently (Ki>300 nM) but has somewhat higher avidity for Mcl1 (Ki∼180 nM).18, 19 These molecules re-engage apoptosis primarily by disrupting endogenous Bcl-2 family interactions and inhibiting sequestration of pro-death BH3 proteins18 although additional mechanisms have been suggested for AT-101.20
NB cells showing a Noxa-dominant profile (e.g. IMR5, NLF) were most sensitive to the Mcl1-neutralizing AT-101 with an IC50 in the 2 μM range and with near-complete cytotoxicity at 5 μM for each; Figure 5a. ABT-737 is a more potent inhibitor of its target proteins, yet higher concentrations were required for similar cytotoxicity in these cell lines, presumably as Mcl1 is not antagonized. On the basis of our mitochondrial profiles, response to ABT-737 may reflect antagonism of Bcl-xL or Bcl-w as BikBH3 is next most potent peptide behind NoxaBH3. In contrast, NB cells with a Bik-dominant response profile (e.g. LAN5, SMS-KCN) were exquisitely sensitive to ABT-737 with IC50s ∼200 nM, two logs less than for AT-101 or for IMR5 sensitivity to ABT-737 (Figure 5b). Therefore, a dominant BikBH3 profile serves as a predictor of exquisite ABT-737 sensitivity and our data suggest this is largely through reduced Mcl1 expression and function.

BH3 profiling predicts NB cell sensitivity to Bcl-2 family small molecule antagonists. 30 000 cells were plated in 96-well plates in duplicate, allowed to settle overnight, and exposed to increasing concentrations of AT-101, ABT-737, or DMSO control. Cell indexes (viable cell number) for duplicate wells are averaged and plotted as a function of time and normalized separately to the cell index reading of each well at the time just before drug addition (t=0). Cells with Mcl1 dependence were more sensitive to AT-101 (a). Cells with a Bik-dominant mitochondrial profile were exquisitely sensitive to ABT-737 (b). Cytotoxicity curves represent the average of at least two separate experiments. Standard deviations between individual experiments were all less than 5% of the values plotted and are therefore not shown. μM, micromolar; nanoM, nanomolar
SK-N-AS and BE2C were insensitive to the highest doses of ABT-737 and had minimal cell death to doses of AT-101 that were cytotoxic to other cell lines (Figure 6). The RPE-1 neural cell control was also completely resistant to ABT-737, as predicted by its BH3 profile, and showed sensitivity to AT-101 comparable to that of NB cell lines with enabler resistant BH3 profiles (Figure 6). Therefore, diversity in mitochondrial responses to BH3 death signals accurately predicts pro-survival Bcl-2 protein addiction patterns within a cell line that can be successfully targeted with small molecule BH antagonists.

NB resistant to enabler BH3s and normal cell controls are resistant to small molecule Bcl-2 family antagonists. RPE-1 is an adherent non-transformed neural cell line. Cytotoxicity curves represent the average of at least two separate experiments. Standard deviations between individual experiments were all less than 5% of the values plotted and are therefore not shown
Discussion
Complexity exists among Bcl-2 protein interactions that mediate mitochondrial apoptosis. Post-translational modifications (PTM), redundancy for death stimuli, and differing affinities among Bcl-2 proteins make expression-based predictions unreliable. Assaying mitochondria for functional responses to BH3 stimuli defined pro-survival addiction patterns that were heterogeneous but reproducible. Clustering analyses defined subsets of NB with an Mcl1 dependence (Noxa-dominant profile), Bcl-xL and/or Bcl-w dependence (Bik-dominant), or generalized resistance to enabler BH3 signals (arising in relapsed tumors). Pro-survival Bcl-2 proteins were heterogeneously expressed and in general did not predict their functional role in apoptosis suppression, although reduced Mcl1 protein expression may be a biomarker for the Bik-dominant cluster. PTM may alter Bcl-2 family functions, such as deamidation of Bcl-xL or phosphorylation of Bcl-2, contributing to discordance between abundance and function. Identifying mitochondrial membrane-resident Bcl-2 family proteins and their PTMs may help discriminate additional biomarkers defining BH3 response clusters.
Not all BH3 peptides were equally efficient at inducing cytochrome c release in NB. Consistent with findings from many laboratories,21 Bid and Bim were most potent in our assays, meeting or exceeding the potency of recombinant tBid. In the ‘direct-activator model’ of mitochondrial apoptosis, the greater activity of Bid and Bim is attributed to their unique ability to physically engage and activate Bak or Bax (as experimentally shown for Bax22). The remaining BH3-only proteins (e.g. Bik and Noxa) are less effective as their affinity is restricted to pro-survival Bcl-2 proteins.7, 8, 23 They enable apoptosis through the neutralization of pro-survival Bcl-2 pockets, but remain dependent on activator BH3 proteins to engage Bak or Bax. Alternatively, the ‘indirect-activator’ model proposes the pro-apoptotic function of BH3-only proteins results from their collective antagonism of the pro-survival Bcl-2 proteins that maintain Bak and Bax in an inactive state.24, 25 The more potent apoptosis-inducing activity of certain BH3 proteins over others is attributed to their broader affinity. Bid and Bim engage a greater number of pro-survival proteins neutralizing their antagonism of Bak and Bax, and increase the likelihood that sufficient Bak or Bax are free to activate apoptosis. Both models are supported by substantial experimental data and are not necessarily exclusive.
Using co-immunoprecipitation we found that Bim was tonically sequestered to pro-survival proteins in NB cells growing at steady state (Figure 3b). Moreover, mitochondria derived from these same cells could be induced to release cytochrome c after exposure to enabler BH3 peptides with restricted affinity for only a subset of Bcl-2 proteins, such as NoxaBH3 for Mcl1 (as with IMR5, Figure 1) suggesting a ‘primed for death’ status consistent with the direct-activator model. Moreover, in a subset of enabler resistant cell lines only putative activator BH3 peptides induced cytochrome c release, whereas combinations of enablers that similarly neutralize all pro-survival members remained ineffectual (data not shown). Indeed, SK-N-AS cells showed little release to enabler BH3 peptides. Yet after exposure to a sublethal etoposide concentration (1 log lower than the IC50), mitochondrial responses to enabler peptides were augmented (Figure 3c) leading to post-treatment SK-N-AS mitochondria clustering with the Noxa-dominant group (Figure 2a).
In our studies, PumaBH3 did not induce robust cytochrome c release, contradictory to our expectations. The relative weakness of PumaBH3 in our assays could not be attributed to an inability to form a stable α-helix (essential for pro-death function,26) as circular dichroism assays demonstrated helicity comparable to other test peptides (data not shown). The peptide sequence has been reported as highly active in similar assays.8 Still, given Puma's non-selective and promiscuous affinity profile (Table 1), it is not likely to have altered our clustering or inferences about pro-survival dependence even were it more active.
Mitochondrial BH3 response profiles accurately predicted whole cell sensitivity to small molecule Bcl-2 family antagonists, and may prove useful in assessing therapeutic strategies targeting chemoresistance in NB. Although AT-101 was more potent against Mcl1-dependent cell lines (Noxa-dominant), there remained modest sensitivity to AT-101 at mid-micromolar concentrations in all cells, including non-transformed cells. Gossypol (the racemic enantiomer of AT-101) is cytotoxic to mouse embryo fibroblasts genetically null for Bax and Bak20 or caspase 9,27 which supports additional cytotoxic effects beyond Bcl-2 family antagonism. The reduced sensitivity of Noxa-dominant NBs to AT-101 (relative to ABT-737 effects against Bik-dominant NB) may be due to its lesser affinity for Mcl1 in comparison to ABT-737 affinity for its targets. ABT-737 activity at micromolar concentrations against Noxa-dominant cells likely reflects a co-dependence on Bcl-xL and Bcl-w suggested by the substantial BikBH3 response. Mcl1 may provide the primary barrier to ABT-737 sensitivity in this subset, as in other cancer models.5, 20, 28, 29 Indeed, our laboratory has shown that siRNA inhibition of Mcl1 in Noxa-dominant cells is cytotoxic and enhances sensitivity to ABT-737 at doses as low as 1 nM.30 Therapeutic opportunities for this group include Mcl1-antagonizing small molecules (AT-101 and others) or combination therapies with ABT-737 and agents that neutralize Mcl1 (e.g. proteosome inhibitors, CDK inhibitors).
Our data showed that ABT-737 was active against NB cells with a Bik-dominant response profile. Previous studies have shown ABT-263, the oral analogue of ABT-737, to be relatively ineffective in a cohort of NB cell lines.31 In that study, the only ABT-737-sensitive cells were NB-1643, which we now show by BH3 response profiling to have a Bik-dominant pattern predicting sensitivity to Bcl-2, -xL, and/or -w antagonism. Immunoblots of cell lines that were exquisitely sensitive to ABT-737 showed a reduction of Mcl1 protein, providing a potential biomarker for this class.
Finally, our studies showed that post-therapy relapsed NBs typically presented mitochondria resistant to enabler BH3 signals. Thus, emergent therapy resistance may be associated with loss of BH3 priming or effectual Bak/Bax engagement and MOMP activation. As Bax and Bak were present, and their mitochondria could be primed to release cytochrome c after etoposide exposure, we presume the apoptotic machinery downstream of Bcl-2 oversight is intact, and that loss of activator BH3 priming is responsible. Comparing BH3 response profiles from cell lines derived from the same patient pre- and post relapse may help define the acquired apoptosis resistance patterns contributing to chemoresistance.
Further studies using available in vivo models are required to validate these findings. As BH3 profiles from fresh xenografts are largely concordant with monolayer culture results, we conclude that the Bcl-2 set point that defines sensitivity to myriad stressors is hardwired in NB cells and suggests that profiling primary tumors may be used to identify tumor-specific therapeutic targets in the Bcl-2 family. We propose that BH3 profiling may be used not only to further our understanding of solid tumor apoptosis deregulation but also to triage NB and other chemoresistant solid tumors for biologically active agents in this class.
Materials and Methods
Cell lines
NB cell lines with MYCN amplification [IMR5,32 NLF, LAN5,33 SMS-SAN, NGP,34 CHP134, SMS-KCN, BE2C, NB-164335] and without [NB6936 and SK-N-AS37] were used. Non-transformed neural RPE1-hTERT and human peripheral T cells were used as controls. Neural cells were grown in RPMI-1640 (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1% OPI, 100 U/ml of penicillin, and 100 mcg/ml gentamicin. T cells were cultured with 10% fetal bovine serum, 10% IL2, 100 U/ml penicillin, and immediately used for mitochondrial assays. Tissue culture was at 37°C in a humidified atmosphere of 5% CO2.
Peptides
Human BH3-only protein sequences were used to design BH3 peptide bioprobes encompassing the α-helical BH3 death domains flanking the consensus L-XXX-GDE motif.9, 14 A summary of previously defined affinities of BH3 peptides for multi-domain Bcl-2 family members obtained by fluorescence polarization assay or surface plasmon resonance are provided in Table 1.7, 8, 14 Peptide sequences used in these experiments, also provided in Table 1, were in most cases identical to those used in the referenced affinity studies. Some peptides were synthesized with an arginine homopolymer (r8) to serve as a protein transduction sequence for whole cell assays38 yet no differential activity in mitochondrial assays was seen between any BH3 peptide and its r8-modified version (Supplementary Figure 1). BidBH3 substituted at two highly conserved residues (BidaltBH3) served as an inactive BH3 control whereas recombinant tBid protein (R&D Systems, Minneapolis, MN, USA) served as a positive control.9 Peptides were synthesized using solid-phase Fmoc chemistry with the N- and C-termini blocked by acetyl and amide groups, respectively, and isolated by HPLC to >95% purity (New England Peptide, Gardner, MA, USA). All working dilutions contained ≤0.8% DMSO, and 1% DMSO in PBS was used as a vehicle control.
Isolation of functional mitochondria
Adherent NB cells were detached with Versene, rinsed thrice in ice-cold PBS, and suspended in a buffer containing 250 mM sucrose, 20 mM HEPES, 1 mM DTT, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 1.5 mM MgCl2 with fresh protease inhibitors (Complete; Roche, Manheim, Germany). For xenograft studies, 105 NB cells were pelleted and resuspended in 0.2 ml of Matrigel. Cell suspensions along with 0.1 ml of air were inoculated subcutaneously into the flanks of nude mice. Exponential tumor growth occurred ∼10 days after inoculation and no treatment was applied to tumors. When tumors were ≤500 mm3 in volume, the mice were killed and the non-necrotic fresh tumors were dissected from their flank. CB17SC-Mscid−/− mice (Taconic Farms, Germantown, NY, USA) were used for NB1643 and SK-N-AS studies and nu/nu athymic mice (Jackson Laboratory, Bar Harbor, ME, USA) for SMS-SAN studies. Tumors were placed in ice cold PBS, disassociated, and filtered through 40 μm sterile nylon mesh filter by gravity and centrifuged. Xenograft cell pellets were resuspended in Tris ammonium chloride and centrifuged. Residual tumor pellets were washed/centrifuged at 1400 r.p.m. twice in cold PBS, with the final pellet re-suspended in mitochondrial isolation buffer with protease inhibitors. Cells were vortexed and incubated on ice before dounce homogenization to rupture membranes. Lysates were serially centrifuged at 4 °C with the supernatant, representing the heavy membrane fraction enriched for mitochondria, collected: 800 × g, 1050 × g, then at 15 000 × g for 10 min each, with mitochondria encompassing the final pellet. A small aliquot was lysed in CHAPS buffer and assessed by optical density for protein concentration. All functional studies were performed with freshly isolated mitochondria.
Cytochrome c release assays
Fresh mitochondria were suspended in 1 M Tris–MOPS, 50 mM KH2PO4, 0.5 M Tris–EGTA, 50 mM glutamate, 0.5 M malate, and 4 M KCl to a final concentration of 1 μg/μl, as described earlier.9 BH3 peptides (50 μM, unless otherwise stated), tBid (10 μM), or 1% DMSO were incubated with mitochondria for 30 min at RT. Solutions were centrifuged at 15 000 × g at 4 °C for 5 min and the supernatant separated. Mitochondria-enriched pellets were lysed in 2% CHAPS buffer and equivalent portions of supernatant and mitochondria were plated in 96-well plates in duplicate for ELISA detection of cytochrome c release according to the manufacturer's protocol (R&D systems). Absolute cytochrome c release for each condition was defined as [supernatant cyto c signal/supernatant+mito cyto c signal] × 100. To control for cytochrome c release from mechanical trauma in processing (that might differ between cell lines), results were scored for % cytochrome c released above the value released in the negative control condition (always <30%). BH3 peptides that caused less cytochrome c release than the negative control, when normalized, had a negative value and were considered inactive. All experiments had biological and technical replicates performed at least twice.
Co-immunoprecipitation
Whole protein lysates (400 μg) were pre-cleared with protein A Agarose beads for 2 h and then incubated with antibody [5 μg of Mcl1 (559027 BD Pharmingen; San Diego, CA, USA), Bcl-2 (M0887, Dako; Glostrup, Denmark), Bcl-xL (Santa Cruz sc-8392), β tubulin (Santa Cruz, Santa Cruz, CA, USA)] followed by Protein A agarose beads. Lysates were centrifuged at 3000 r.p.m. at 4°C and repeatedly washed. IP/bead complex was boiled, centrifuged, and immunoblotted for Mcl1 (Dako), Bcl-2 (Alexis Biochemicals, Plymouth Meeting, PA, USA), β tubulin, or Bcl-xL to confirm the protein was successfully isolated, then with tBid, Bak (Santa Cruz) and Bim (Axxora, San Diego, CA, USA), Puma (Santa Cruz), and Bad (Cell Signaling, Danvers, MA, USA) to assess for pro-survival protein interactions/binding partners.
Immunoblots
Cells were cultured in replete media with 10% FBS, collected with Versene, washed in PBS, and lysed on ice in a 1% NP40 buffer with fresh protease inhibitors (Roche Complete). Protein (25 μG) was electrophoresed through a Tris-Glycine gel, and immunoblotted using antibodies to: β-tubulin, Bak and Mcl1 (Santa Cruz), Bcl-w (Alexis), Bax and Bcl-xL (Cell Signaling), Bcl-2 (Dako), and A1 (Abcam; Cambridge, MA, USA).
Cytotoxicity assays
A label-free cell electronic sensoring system (RT-CES, Roche/ACEA Biosciences, San Diego, CA, USA) measured impedance in real-time to obtain a cell index of adherent cell biomass. This assay is label free and allows real time biomass monitoring.39, 40 Cells were seeded into multi-well sensor microplates and allowed to adhere overnight. Drug was added during exponential growth and cell index monitored over 72 h. AT-101 (Ascenta Therapeutics) and ABT-737 (Abbot Laboratories) were studied at a range of concentrations up to 20 μM; 1% DMSO was used as a vehicle control. IC50 was calculated using a logarrhythmic 4-parameter curve fitting program through Prism4 software analysis (Graphpad).
Statistical analyses
NB responses to BH3 peptides were analyzed by unsupervised hierarchical clustering with Ward's minimum variance method. All data were averaged from both biological and technical replicate experiments for each cell line before clustering. R-square and root mean square standard deviation were used to determine the number of clusters. All other comparisons to test for significance used the two-tailed Student's t-test.
Abbreviations
- NB:
-
neuroblastoma
References
Walensky LD . BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ 2006; 13: 1339–1350.
Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A . Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 2007; 117: 112–121.
Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A . BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood 2008; 111: 2300–2309.
Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A . BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007; 12: 171–185.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10: 375–388.
Reed JC . Apoptosis-targeted therapies for cancer. Cancer Cell 2003; 3: 17–22.
Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006; 9: 351–365.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 2005; 17: 525–535.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ . Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002; 2: 183–192.
Moreau C, Cartron PF, Hunt A, Meflah K, Green DR, Evan G et al. Minimal BH3 peptides promote cell death by antagonizing anti-apoptotic proteins. J Biol Chem 2003; 278: 19426–19435.
Fulda S, Susin SA, Kroemer G, Debatin KM . Molecular ordering of apoptosis induced by anticancer drugs in neuroblastoma cells. Cancer Res 1998; 58: 4453–4460.
Poulaki V, Mitsiades N, Romero ME, Tsokos M . Fas-mediated apoptosis in neuroblastoma requires mitochondrial activation and is inhibited by FLICE inhibitor protein and Bcl-2. Cancer Res 2001; 61: 4864–4872.
Hogarty MD . The requirement for evasion of programmed cell death in neuroblastomas with MYCN amplification. Cancer Lett 2003; 197: 173–179.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403.
Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001; 293: 1829–1832.
Leung KT, Li KK, Sun SS, Chan PK, Ooi VE, Chiu LC . Activation of the JNK pathway promotes phosphorylation and degradation of BimEL – a novel mechanism of chemoresistance in T-cell acute lymphoblastic leukemia. Carcinogenesis 2008; 29: 544–551.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005; 19: 1294–1305.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA 2000; 97: 7124–7129.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Letai AG . Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer 2008; 8: 121–132.
Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG et al. BAX activation is initiated at a novel interaction site. Nature 2008; 455: 1076–1081.
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 2006; 8: 1348–1358.
Uren RT, Dewson G, Chen L, Coyne SC, Huang DC, Adams JM et al. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J Cell Biol 2007; 177: 277–287.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007; 315: 856–859.
Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ . BID: a novel BH3 domain-only death agonist. Genes Dev 1996; 10: 2859–2869.
Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ 2009; 16: 1030–1039.
Hallaert DY, Spijker R, Jak M, Derks IA, Alves NL, Wensveen FM et al. Crosstalk among Bcl-2 family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual exhaustion of Bcl-2 protection. Cell Death Differ 2007; 14: 1958–1967.
Chen S, Dai Y, Harada H, Dent P, Grant S . Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 2007; 67: 782–791.
Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther 2009; 8: 1587–1595.
Lock R, Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer 2008; 50: 1181–1189.
Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE . Definition of a continuous human cell line derived from neuroblastoma. Cancer Res 1970; 30: 2110–2118.
Reynolds CP, Biedler JL, Spengler BA, Reynolds DA, Ross RA, Frenkel EP et al. Characterization of human neuroblastoma cell lines established before and after therapy. J Natl Cancer Inst 1986; 76: 375–387.
Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983; 305: 245–248.
Schlesinger HR, Gerson JM, Moorhead PS, Maguire H, Hummeler K . Establishment and characterization of human neuroblastoma cell lines. Cancer Res 1976; 36: 3094–3100.
Feder MK, Gilbert F . Clonal evolution in a human neuroblastoma. J Natl Cancer Inst 1983; 70: 1051–1056.
Schmechel D, Marangos PJ, Brightman M . Neuron-specific enolase is a molecular marker for peripheral and central neuroendocrine cells. Nature 1978; 276: 834–836.
Goldsmith KC, Liu X, Dam V, Morgan BT, Shabbout M, Cnaan A et al. BH3 peptidomimetics potently activate apoptosis and demonstrate single agent efficacy in neuroblastoma. Oncogene 2006; 25: 4525–4533.
Solly K, Wang X, Xu X, Strulovici B, Zheng W . Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev Technol 2004; 2: 363–372.
Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008; 455: 930–935.
Acknowledgements
ABT-737 and AT-101 were generously provided by Abbott Laboratories and Ascenta Therapeutics, respectively. We thank Steven Elmore from Abbott Laboratories and Lance Leopold from Ascenta Therapeutics for their direction and assistance with drug usage. We thank David Teachey for provision of T cells, Josh Courtright for xenograft assistance, and Anthony Letai for help in optimizing mitochondrial isolation assays. This work was supported by NIH CA97323, the King Family, the Richard and Sheila Sanford Chair in Pediatric Oncology, and the Alex's Lemonade Stand Foundation (to MDH); and NIH K08-CA128925, The Caitlin Robb Foundation, and Hope Street Kids Foundation (to KCG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by V De Laurenzi
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Goldsmith, K., Lestini, B., Gross, M. et al. BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ 17, 872–882 (2010). https://doi.org/10.1038/cdd.2009.171
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.171
Keywords
This article is cited by
-
Targeting the apoptosis pathway to treat tumours of the paediatric nervous system
Cell Death & Disease (2022)
-
A direct comparison of selective BH3-mimetics reveals BCL-XL, BCL-2 and MCL-1 as promising therapeutic targets in neuroblastoma
British Journal of Cancer (2020)
-
Rationally derived drug combinations with the novel Mcl-1 inhibitor EU-5346 in breast cancer
Breast Cancer Research and Treatment (2019)
-
Too many targets, not enough patients: rethinking neuroblastoma clinical trials
Nature Reviews Cancer (2018)
-
Select Bcl-2 antagonism restores chemotherapy sensitivity in high-risk neuroblastoma
BMC Cancer (2016)

{kind=link}