
Abstract
Apoptosis is a conserved homeostatic process critical for organ and tissue morphogenesis, development, and senescence. This form of programmed cell death also participates in the etiology of several human diseases including cancer, neurodegenerative, and autoimmune disorders. Although the signaling pathways leading to the progression of apoptosis have been extensively characterized, recent studies highlight the regulatory role of changes in the intracellular milieu (permissive apoptotic environment) in the efficient activation of the cell death machinery. In particular, glutathione (GSH) depletion is a common feature of apoptotic cell death triggered by a wide variety of stimuli including activation of death receptors, stress, environmental agents, and cytotoxic drugs. Although initial studies suggested that GSH depletion was only a byproduct of oxidative stress generated during cell death, recent discoveries suggest that GSH depletion and post-translational modifications of proteins through glutathionylation are critical regulators of apoptosis. Here, we reformulate these emerging paradigms into our current understanding of cell death mechanisms.
Similar content being viewed by others

Main
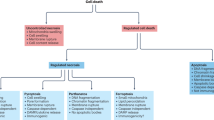
Apoptosis or programmed cell death is a ubiquitous homeostatic process involved in numerous biological systems. Under physiological conditions it is critical not only in the turnover of cells in tissues but also during normal development and senescence. Moreover, its deregulation has been widely observed to occur as either a cause or consequence of distinct pathologies including cancer, autoimmune, and neurodegenerative diseases. Apoptosis is a highly organized program induced by a myriad of stimuli that are characterized by the progressive activation of precise pathways leading to specific biochemical and morphological alterations in individual cells without involving an inflammatory response. Early stages of apoptosis are characterized by initiator caspase activation, cell shrinkage, loss of plasma membrane lipid asymmetry, and chromatin condensation. The execution phase of apoptosis is characterized by activation of executioner caspases and endonucleases, apoptotic body formation, and cell fragmentation1 (Figure 1). Interestingly, recent studies have shown that changes in the intracellular milieu of the cells, such as alterations in the redox environment, are important regulators of the progression to apoptosis.2 These discoveries have lead to the concept of a permissive apoptotic environment necessary for the activation of the proper signaling pathways regulating apoptosis. Glutathione (GSH) depletion is an early hallmark in the progression of cell death in response to a variety of apoptotic stimuli in numerous cell types3, 4 (Figure 2), although the exact role of GSH depletion in apoptosis is still controversial. We review recent data that show new insights into the understanding of the causes and consequences that alterations in the redox environment of the cell have on apoptosis.

Apoptotic signaling pathways (see text for further details)

GSH depletion is a hallmark of apoptosis. Apoptosis was induced in the human lymphoma cell line (Jurkat) by activation of Fas death receptors (FasL), stress (UV radiation), or cytotoxic drugs. Briefly, Jurkat cells were exposed for 8 h to either 25 ng/ml FasL, 15mJ/cm2 UVC radiation, 35 μM cycloheximide, 100 μM etoposide, or 20 nM staurosporine. GSH content and cell viability was assessed by FACS using a BD-LSRII and BD-FACSDiva software. Cells were preloaded with monochlorobimane (a GSH-binding dye; 10 μM, 15 min) and propidium iodide (PI) (for plasma membrane integrity; 10 μg/ml, 5 min) before FACS analysis; 10 000 cells per sample were analyzed and results are represented as histograms or contour plots showing the distribution of cells with differences in GSH content and cell viability. For further details refer to Ref.20, 21 Apoptosis induces a decrease in the population of cells with high intracellular GSH content (GSHi) with a concomitant increase in the populations of cells with depleted GSHi. Two populations of cells with depleted GSH content can be identified based on other biochemical characteristics. The first decrease in GSHi is associated with the initiation phase of apoptosis, whereas the secondary decrease in GSHi is observed after the execution phase is triggered (upper panels).20, 21 GSH depletion is an early event during apoptosis, which occurs before the loss of cell viability, measured by PI uptake (lower panels)
The Role of GSH in the Regulation of Apoptosis
GSH synthesis, depletion, and apoptosis
GSH (L-γ-glutamyl-L-cysteinyl-glycine) in its reduced form is a tripeptide enzymatically formed by glycine, cysteine, and glutamate, and is the most abundant non-protein thiol in mammalian cells. GSH acts as a reducing agent and as a major antioxidant within cells by maintaining a tight control of the redox status. GSH is also involved in many distinct physiological reactions including cellular signaling, metabolism of xenobiotics, thiol disulfide exchange reactions, and as an important reservoir of cysteine. GSH is a ubiquitous molecule that is produced intracellularly attaining mM concentrations whereas plasma concentrations of GSH are at low μM levels because of its rapid catabolism. GSH is 85–90% freely distributed in the cytosol, but can also be compartmentalized in organelles including the mitochondria, the peroxisomes, the nuclear matrix, and the endoplasmic reticulum (ER) after its cytosolic synthesis. Synthesis of GSH is initiated by the synthesis of γ-glutamylcysteine from glutamate and cysteine by the γ-glutamylcysteine synthetase (γ-GCS). Glycine is subsequently added by the activity of GSH synthetase (GS).4, 5
GSH is essential for cell survival as the GSH-depleted knock-out mouse of γ-GCS dies from massive apoptotic cell death.6 As the availability of cysteine is the rate-limiting factor in GSH formation, cysteine starvation has been shown to induce apoptosis by GSH depletion.7 Several studies have shown a correlation between GSH depletion and the progression of apoptosis, and knockdown of γ-GCS induces time-dependent apoptosis in various cell types.8 Conversely, overexpression of γ-GCS protects against apoptosis induced by stimuli that activate both extrinsic and intrinsic cell death pathways.9, 10 γ-GCS is a heterodimer of a catalytic subunit and a regulatory subunit. The γ-GCS promoter contains many potential cis-acting elements, including consensus recognition sites for binding of activator protein-1 (AP-1 or TRE), activator protein-2 (AP-2), Sp-1, nuclear factor κ-B (NF-κB), and the electrophile responsive element EpRE (also called the antioxidant response element). EpREs are located in the 5′-flanking sequences of both subunit genes and are thought to mediate gene induction following xenobiotic exposure.11 The EpRE is the cis-acting DNA element that mediates the NF-E2-related factor-2 (Nrf2)-dependent regulation of gene expression.12 Downregulation of Nrf2 has been shown to reduce γ-GCS gene expression and decrease GSH levels13 and GSH-dependent enzymes.14 Interestingly, Nrf2 levels have been directly associated with resistance to apoptosis.15 Nrf2 has also been shown to protect against oxidative stress-induced cell death by upregulation of enzymes involved in GSH biosynthesis, use, and export.16
High intracellular GSH levels have been associated with apoptotic resistant phenotypes in several models of apoptosis,17, 18 and GSH depletion by itself has been observed to either induce or stimulate apoptosis.17, 19 For example, buthionine sulfoximine (BSO)-induced GSH depletion does not trigger apoptosis, but potentiates death receptor-induced apoptosis in T cells.17, 19 GSH supplementation with N-acetyl-L-cysteine (NAC) also prevents apoptosis20, 21, 22 Replenishment of intracellular GSH pools with sulfur amino acids including S-adenosyl-methionine, which is a precursor of GSH through its sequential conversion from S-adenosylhomocysteine (SAH) to cysteine, has also been shown to protect against apoptosis.18, 23
At present, the precise contribution of cytosolic versus mitochondrial GSH pools in apoptosis is not fully understood. Some reports suggest that apoptosis correlates directly with depletion of cytosolic GSH rather than with mitochondrial GSH depletion .24 However, other studies have shown that mitochondrial GSH depletion is important in triggering the cell death cascade. Accordingly, promotion of cell death by diseases or treatments that deplete cellular GSH seems to correlate closely to the extent of depletion of mitochondrial GSH rather than the changes in the cytoplasmic pool.25, 26 Mitochondrial thiols have been clearly shown to act as regulators of apoptosis.27 Selective depletion of mitochondrial GSH sensitizes cells to oxidative stress- and nitrative stress-induced apoptosis.28 Mitochondria depend on GSH for the prevention of oxidative damage because of aerobic respiration. Salvage of glutathione-disulfide (GSSG) by GSH reductase (GR) and the uptake of cytosolic GSH across the outer membrane are required for the maintenance of mitochondrial GSH levels because these organelles are incapable of de novo synthesis. It has also been proposed that GSH must be actively transported into the mitochondria or exchanged for another anion. This observation likely reflects the fact that the matrix space of the mitochondria possesses a negative membrane potential relative to the cytoplasm, and also that GSH is a negatively charged molecule at physiological pH. Some of the possible candidates involved in the mitochondrial import of cytosolic GSH include the monocarboxylate, dicarboxylate, 2-oxoglutarate, tricarboxylate, glutamate-hydroxide, and glutamate-aspartate transporters.25 Accordingly, overexpression of the mitochondrial glutathione transporters (dicarboxilate and 2-oxoglutarate carriers) has been shown to protect cells from chemically induced apoptosis.29, 30 Ethanol toxicity has been shown to deplete mitochondrial GSH by inhibition of its uptake from the cytosol, which potentiates acetaminophen- and tumor necrosis factor (TNF-α)-induced apoptosis.31, 32 Recently, inhibition of mitochondrial GSH transport has been reported to exacerbate oxidant-induced cytochrome c (Cyt c) release, caspase 9 activation, and apoptosis.33
Intracellular GSH depletion is an early hallmark in the progression of cell death in response to different apoptotic stimuli3, 4 (Figure 2). GSH depletion during apoptosis induced by cytotoxic agents, which by themselves induce oxidative stress such as xenobiotics, chemotherapeutics, and metals, has been reported to be mediated by GSH oxidation to GSSG by reactive species (RS) of both oxygen (ROS) and nitrogen (RNS), or by its conjugation to highly reactive compounds34, 35 (Figure 3). In contrast, apoptosis induced by distinct stimuli, such as activation of death receptors, has been reported to be mediated a plasma membrane efflux transport of GSH.21 Inhibition of GSH depletion under these conditions is able to rescue cells from apoptosis.21 Several studies have suggested that both multidrug resistance proteins (MRP)36 and/or GSH/organic anion (OA−) exchangers might be involved in GSH efflux during apoptosis.21 The MRPs act as transporters of GSH, GSSG, and GSH-adducts and require the hydrolysis of ATP for its transport activity.37 Pharmacological activation of MRPs induces apoptosis by GSH depletion.38 In clear contrast, other studies have shown that inhibition of MRP-mediated transport accelerates apoptosis.20, 21 Conversely, overexpression of MRP proteins inhibit cell death induced by death receptor activation and cytotoxic drugs.39 These data suggest that MRP-mediated transport might act in a protective manner against apoptosis. The organic anion transporting polypeptides (OATP) have also been proposed to mediate GSH efflux by a GSH/OA− exchange,40 although recent studies have suggested that GSH/OA− exchange is not mediated by this family of transporters.41 Thus, there is a strong possibility that GSH efflux is mediated by a different and still uncharacterized entity. Whether GSH is depleted by its efflux, intracellular oxidation, or conjugation seems to depend on the nature and dose of the apoptotic stimuli. Interestingly, recent findings have suggested that γ-GCS is a direct target of caspase 3,42 which may represent another mechanism for preventing GSH replenishment ensuring apoptosis. The cystic fibrosis transmembrane conductance regulator has also been suggested to mediate transport of GSH during apoptosis, which modulates the activation of Bax.43 Connexins, which when unapposed form hemichannels, have been recently suggested to mediate the efflux of GSH but their role in the progression of apoptosis has not been studied yet.44

Mechanisms involved in GSH depletion during apoptosis. GSH depletion during apoptosis can occur by distinct mechanisms. (a) Activation of death receptors induces GSH loss by its extrusion across the plasma membrane through GSH transporters or pumps (GSH-T). Recent evidence suggests that caspases might be modulating this process. (b) Activation of intrinsic pathways of apoptosis might mediate GSH depletion by its oxidation to GSSG because of the generation of reactive species (RS) of both oxygen (ROS) and nitrogen (RNS) that arise from impairment of mitochondrial function. A crosstalk between transport- and RS-mediated GSH depletion might exist according to the apoptotic conditions. (c) Apoptotic signals arising from pro-oxidant stimuli mediate GSH depletion by both its direct oxidation and/or conjugation. (d) GSSG and GSH-adducts formed are further reduced or extruded to avoid their deleterious effects inside the cell. Other mechanisms such as impairment of GSH synthesis and recycling might also contribute to preventing GSH replenishment during apoptosis
GSH depletion by its efflux has been thought to regulate intracellular apoptotic signaling, However, it has also been suggested that efflux of GSH and/or its extracellular accumulation might regulate apoptosis by different signaling events mediated by GSH pro-oxidant catabolism through the γ-glutamyl transpeptidase (γ-GT).45, 46 The γ-GT, expressed mainly on the apical surface of cells, initiates the catabolism of not only GSH, but of glutathione S-conjugates and glutathione-complexes by removal of the γ-glutamyl moiety from GSH and GSH-conjugated compounds, producing cysteinylglycine or cysteinylglycine-conjugates. These products are subsequently hydrolyzed by ectoprotein dipeptidases and then, cysteine, together with the γ-glutamyl-amino acids formed, are uptaken by the activity of specific transporters. Once in the cytosol, γ-glutamyl-derivatives are substrates for the γ-glutamyl cyclotransferase that forms 5-oxoproline, which is finally converted to glutamate by the activity of the 5-oxoprolinase.46 Thus, by cleavage of GSH and the subsequent recapture of cysteine, cystine, and γ-glutamyl-amino acids, γ-GT facilitates replenishment of intracellular GSH pools. Accordingly, overexpression of γ-GT has been shown to protect cells from apoptosis induced by oxidative stress.47
GSH and apoptotic signaling cascades
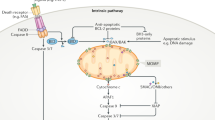
The signaling cascades that regulate the progression of apoptosis have been extensively studied and characterized; and both extrinsic and intrinsic pathways have been described for the activation of apoptosis (Figure 1). Induction of apoptosis through extrinsic pathways is triggered by death receptors such as those activated by Fas ligand or FasL (Fas, CD95/Apo-1); by TNF-related apoptosis-inducing ligand or TRAIL (DR4, DR5); and by TNF-α (TNFR1). Activation of CD95, DR4, and DR5 leads to the formation of the death-inducing signaling complex (DISC) through the recruitment of the Fas-associated death domain (FADD), caspase 8 (and in some cases caspase 10), and the cellular FLICE-inhibitory protein (FLIP). Initiator caspase 8 is activated and further amplifies the apoptotic cascade by activation of executioner caspases (3, 6, and 7). In cells that have lower levels of DISC formation and thus, reduced caspase 8 activation (Type II cells), the progression of apoptosis relies on an amplification loop induced by caspase 8-dependent cleavage of the Bcl-2 family protein, Bid, translocation to the mitochondria, and subsequent release of Cyt c. In contrast, TNFR1 signaling results in the formation of two signaling complexes. TNF-induced complex I formation mediates the recruitment of receptor-interacting protein, TRADD (TNFR-associated death domain protein), and TRAF-1/2 (TNFR-associated factor). This complex lacks FADD and procaspase 8, but it is reported to translocate to the cytosol in which FADD, caspase 8/10, and FLIP are recruited to form the traddosome or complex II in which activation of caspase 8 takes place. In addition, complex I has been shown to mediate other pro-apoptotic signaling cascades including the activation of the apoptosis signal-regulating kinase 1, and the stress activated kinase (SAPK) JNK leading to the transcriptional/post-transcriptional regulation of apoptotic genes.1
The intrinsic pathways of apoptosis involve the mitochondria, the ER, and the DNA damaging pathways. These pathways are activated by a wide variety of stimuli including chemotherapeutic and cytotoxic agents (environmental pollutants, xenobiotics, drugs), stress (radiation, hyperglycemia, hypoxia, oxidative, and osmotic stress), and cytokine withdrawal. Activation of the mitochondria pathway mediates the release of Cyt c that is associated with the opening of the mitochondrial permeability transition pore (MPTP) and loss of the mitochondrial membrane potential. Although MPTP was initially proposed to be formed by the voltage-dependent anion channel, the adenine-nucleotide translocator, and the cyclophilin D, recent studies have suggested that mitochondrial apoptosis is independent of these complex, which in contrast might be involved in cell death by necrosis. The intrinsic mitochondrial pathway is regulated by Bcl-2 family proteins. The BH3-only proteins (members of the Bcl-2 family) Bad, Bid, Bim, NOXA, and PUMA regulate the anti-apoptotic Bcl-2 proteins (Bcl-2 and Bcl-xl) to promote apoptosis. Bcl-2 and Bcl-xl inhibit Bax and Bak. Induction and/or activation of BH3-only proteins de-repress Bax and Bak by direct binding and inhibition of the Bcl-2 anti-apoptotic family members. Bax and Bak are crucial for inducing the permeabilization of the outer mitochondrial membrane and the release of Cyt c. This leads to the recruitment of Apaf1 into the apoptosome and activates caspase 9 to further regulate execution caspases.1
GSH depletion has been shown to regulate both extrinsic and intrinsic apoptotic signaling cascades at distinct checkpoints. GSH depletion can predispose cells to apoptosis or directly trigger cell death by modulation of both the permeability transition pore formation and the activation of execution caspases.48, 49 In vitro studies have shown that a reduction in the GSH content is necessary for the formation of the apoptosome.50 GSH depletion activates the intrinsic apoptotic pathway initiator Bax by its oxidation-dependent dimerization.51 GSH depletion has also been shown to trigger Cyt c release and it has been proposed that released Cyt c from the mitochondria needs to be oxidized for its pro-apoptotic action, which would require cytosolic GSH levels to be depleted.52 Recently, it was shown that in healthy neurons and cancer cells, Cyt c is reduced and held inactive by increased GSH content, generated as a result of glucose metabolism through the pentose phosphate pathway.53 TNF-α-induced apoptosis has also been shown to depend on caspase-dependent GSH depletion that further regulates ceramide production by the neutral sphingomyelinase.54 In addition, GSH depletion has also been shown to impair TNF-α-induced NF-κB signaling sensitizing cells to apoptosis.55
The anti-apoptotic role of Bcl-2 has been widely linked to GSH content. Bcl-2 has been shown to regulate GSH content in different cellular compartments.56 Overexpression of Bcl-2 increases GSH levels and inhibits mitochondrial-induced cell death elicited by GSH-depleting reagents.57 Recent studies suggest that Bcl-2 can regulate the mitochondrial GSH pool by a direct interaction with GSH through the BH3 groove, which mediates its antioxidant function.58 Accordingly, depletion of intracellular GSH has been reported to overcome Bcl-2-mediated resistance to apoptosis.48 However, these effects seem to be cell type-specific and context-dependent.59 The anti-apoptotic effect of Bcl-xl has also been attributed to the regulation of GSH homeostasis by preventing GSH loss.60
Other intrinsic pathways of apoptosis are also regulated by GSH homeostasis. ER stress is induced by alterations in cellular energy levels, the redox state, or Ca2+concentration, which results in the accumulation and aggregation of unfolded proteins. Severe ER stress has been proposed to lead to the activation of JNK kinase and induction of the C/EBP homologous protein (CHOP), which impairs the anti-apoptotic function of Bcl-2, leading to the activation of Bim, Bax, and Bak. Activation of these pro-apoptotic Bcl-2 family members transmits the apoptotic signal from the ER to the mitochondria leading to the execution of death by activation of caspases and/or ER-mediated Ca2+ overload. ER stress has also been proposed to lead to the activation of caspase 12/4 but this observation is still controversial. In addition, a role for caspase 2 and ASK-1 in ER stress-induced apoptosis has been proposed.1 ER stress-induced apoptosis has been shown to trigger oxidative stress and to mediate GSH depletion by its oxidation.61 The lumen of the ER contains a relatively high concentration of GSSG, which is maintained in part by the selective transport of GSH from the cytosol. This gradient allows the formation of disulfide bonds and their isomerization through the activity of protein disulfide isomerases (PDI) and oxidoreductases. Reduced PDI is further oxidized by the flavoprotein Ero1 that generates ROS as byproducts, which are then detoxified by GSH, thereby generating high levels of GSSG. PDI can be either reduced by GSH or oxidized by GSSG. Finally, high luminal GSSG concentrations are then secreted from the ER.62, 63 Thus, GSH serves as a redox buffer against ROS generated in the ER. The accumulation of unfolded proteins in the ER has been recently shown to induce PERK-dependent activation of Nrf2 that contributes to the maintenance of GSH levels. Enhanced GSH content prevents the accumulation of ROS during the unfolded protein response. Thus perturbations in cellular GSH concentrations have been shown to sensitize cells to ER-induced apoptosis.64 ER stress also induces CHOP expression, which has been shown to promote GSH depletion and oxidative injury that is counteracted by the action of Bcl-2.65
Specific DNA lesions or damage, including cross-links and double-strand breaks (DSBs) are well known to trigger apoptosis. DSBs are detected by ataxia telangiectasia mutated gene, ataxia telangiectasia, and Rad3-related proteins, which signal downstream to the CHK1 and CHK2 checkpoint kinases, and p53. p53 induces transcriptional activation of pro-apoptotic factors such as FAS, PUMA, and BAX. Apoptosis on DNA damage has also been shown to be mediated by activation of caspase 2 and JNK.1 Oxidative DNA damage and impaired cellular repair mechanisms contribute to apoptosis. Recent reports have suggested a critical role of nuclear GSH pools in protecting DNA from oxidative modifications that may alter the efficiency of the DNA repair machinery. The nuclear GSH pool is thought to be maintained by the diffusion of GSH into the nucleus across nuclear pores.66, 67 Conjugation of GSH is an essential aspect of xenobiotic and normal physiological metabolism. The glutathione S-transferases (GST) form a class of enzymes with overlapping substrate specificities that generate a large set of thioesthers, called glutathione S-conjugates. GSTs can also serve as peroxidases, isomerases, and thiol transferases. GSTs protect against DNA damage-induced apoptosis by scavenging the formation of lipid-peroxide-modified DNA on oxidative stress.68 Mitochondrial DNA damage is also modulated by GSH which reduces its susceptibility to oxidant-induced damage and apoptosis.69 DNA cross-linker agents, such as cisplatin, have been reported to induce apoptosis paralleled by GSH depletion. In addition, apoptosis induced by the DNA intercalating agent doxorubicin was shown to be potentiated by BSO-induced GSH depletion.17
Redox signaling in apoptosis by GSH
As summarized above, GSH content and its depletion have been widely reported to regulate a variety of apoptotic signaling pathways, however, the precise molecular mechanisms involved in this process are still unclear. Changes in the intracellular thiol-disulfide (GSH/GSSG) balance are considered major determinants in the redox status/signaling of the cell.70 GSH constitutes the major intracellular antioxidant defense against RS and oxidative stress. GSH has been shown to scavenge directly a wide variety of RS, including superoxide anion (•O2−), hydroxyl radical (•OH−), singlet oxygen (1O2), protein-, and DNA radicals, by donating electrons and becoming oxidized to thyil radical (GS•). The generation of disulfide bonds between two GS• leads to the further formation of GSSG. GSH also catalytically detoxifies cells from peroxides such as hydroperoxides (H2O2), peroxynitrite (OONO−), and lipid peroxides (LOO•) by the action of GSH peroxidases (GPX) and peroxiredoxins (PXR). Accumulation of GSSG on oxidative stress has been observed to be toxic to the cell. GSSG has been shown to directly induce apoptosis by the activation of the SAPK/MAPK pathway.71 An early and transient rise in intracellular GSSG has been shown to precede Cyt c release and caspase 3 activation during apoptosis. In these studies GSH/GSSG ratio was observed to return to normal levels 1 h after the apoptotic stimuli, however, it did not influence the progression of cell death.72 GSSG is reduced back to GSH by the action of GR, which requires reduced nicotinamide adenine dinucleotide phosphate (NADPH) as an electron donor reductant. Under physiological conditions GSSG is maintained at ∼1% of the total cellular glutathione concentration and increases in GSSG are counteracted by its rapid reduction by GR. GR requires reduced NADPH as the electron donor reductant. Glucose-6-phosphate dehydrogenase (G6PD), the first and rate-limiting enzyme of the oxidative pentose phosphate cycle, is indispensable for the regeneration of NADPH from NADP+73. G6PD also helps in the maintenance of protein mixed disulfides (protein-SSG) in the cell.74 Impairment of G6PD sensitizes cells to oxidant-induced apoptosis.75 GSH depletion during apoptosis is also paralleled by decreases in NADPH availability, which might contribute to a sustained cellular redox imbalance by impairing GSSG reduction by GR.76 Increased NADPH formation through the penthose pathway has been shown to protect against apoptosis induced by excitotoxicity, death receptors, and serum deprivation.77, 78 Inhibition of GR has also been shown to induce GSH depletion and oxidative stress, which sensitizes cells to undergo apoptosis.79 In addition, GSSG has also been shown to be detoxified by its efflux transport across the plasma membrane.80
Cellular stress has been widely reported to induce apoptosis primarily through oxidant toxicity in which depletion of GSH by its oxidation has been considered to be just a byproduct of oxidative stress. As GSH depletion has been shown to sensitize cells to agents that induce oxidative cell death, apoptosis under these conditions has been assumed to be largely regulated by the signaling action of RS of oxygen and nitrogen. Indeed the role of RS in apoptosis has been extensively studied,81 and several GSH-dependent antioxidant enzymes protect cells from undergoing apoptosis. Glutathione peroxidases (GPX) are selenoproteins that reduce peroxides with different substrate specificity. GPX1 whose preferred substrate is hydrogen peroxide is the most abundant isoform found in the cytoplasm of nearly all mammalian tissues whereas GPX2 is an intestinal and extracellular enzyme.82 GPX1 and GPX2 have been reported to protect against apoptosis induced by oxidative stress83, 84 ischemia/reperfusion injury85 and doxorubicin86 and to reduce pro-apoptotic Bax expression.87 GPX4 (also known as phospholipid hydroperoxide glutathione peroxidase, PHGPX) overexpression has been reported to protect against oxidative stress-induced apoptosis.88 GPX4 also protects against oxidative stress and amyloid-induced apoptosis.89 More recently, GPX4 downregulation has been shown to induce 12/15 lipoxygenase-dependent lipid peroxidation and apoptosis-inducing-factor-mediated cell death.90, 91 Overexpression of the mitochondrial GPX4 was also shown to protect against apoptosis induced through the intrinsic mitochondrial pathway by reducing mitochondrial hydroperoxide accumulation.92
The role of GSH depletion and ROS in apoptosis induced by physiological stimuli such as death receptor activation, which does not induce oxidative stress by itself, remains more controversial.93 GSH depletion has been shown to occur at earlier stages during the progression of the cell death program and it is followed by a delayed accumulation of ROS, which requires GSH depletion.20 Several reports have shown contradictory roles including both inhibitory and stimulatory effects of ROS on apoptosis.2, 94 As GSH is the major antioxidant within the cells, it has been largely thought that GSH regulates apoptosis by preventing the accumulation of ROS. GSH peroxidase (GPX) has been shown to protect against apoptosis induced by Fas activation.95 However, death receptor (Fas and TNF)-induced cell death was shown to be similar in animals deficient in GPX compared with WT.96 Many studies have linked ROS formation with the progression of Fas-induced apoptosis by the protective effect of extracellular thiols such as GSH and NAC, effects ascribed to their known capacities as antioxidants.94 However, protective effects of thiol compounds on apoptosis in the absence of excessive ROS formation are also observed.97 Additionally, apoptosis has been suggested to occur under anaerobic conditions in the absence of ROS formation,98 or in cells lacking mitochondrial DNA, which are deficient in respiration and mitochondrial oxidative phosphorylation, the source of ROS during apoptosis.99 These results imply a more active and widespread role for GSH in the regulation of apoptosis rather than simply acting as an antioxidant/scavenger against oxidative stress and ROS. GSH depletion seems to be required for ROS formation during apoptosis. For example, we and others have recently shown that GSH depletion is necessary for the generation of ROS during FasL-induced apoptosis,20 and specifically observed that GSH content, but not the excess in ROS formation and oxidative stress, regulates apoptosis induced by Fas activation.20 Other studies have also shown that apoptosis seems to be actively regulated by GSH content and not by excessive oxidative stress and ROS generation.100 These findings support the idea of a direct role of GSH in the regulation of the apoptotic machinery independent from oxidative stress. Perhaps, excessive ROS overload might well be just an epiphenomena associated with the depletion of GSH.
Protein glutathionylation and apoptosis
Recently, oxidative post-translational modifications (OPMs) in proteins have been shown to regulate the activity of a wide variety of proteins. OPMs in general can be classified as reversible and irreversible modifications. Reversible modifications include cysteine residues modified through alternative redox-based modifications (nitrosylation, SNO; hydroxylation, SOH; glutathionylation, SSG; disulfide bond formation, S–S) that may enable differential effects on protein function. Protein (S-) glutathionylation (protein-SSG, also known as [S-]glutathiolation) refers to the formation of a protein mixed disulfide between the cysteine of GSH and a cysteine moiety of a protein (thiol exchange reactions). One of the most accepted mechanisms of protein-SSG formation is the reaction of GSSG with protein sulfhydryls (given by exposed protein cysteine residues). Under non-stressed conditions, the intracellular environment favors a fully reduced thiol state. It has been shown that a significant basal level of protein-SSG (∼1%) is found within cells, which is proportionally upregulated by increases in GSSG content. S-glutathionylation of proteins has a dual role in protecting against irreversible protein thiol oxidation and in regulating protein function.101 Oxidative stress and ROS formation during apoptosis induced by pro-oxidant conditions might regulate protein-SSG formation. However, depletion of GSH in response to non-pro-oxidant apoptotic stimuli (activation of death receptors) and as a consequence, a decrease in the GSH/GSSG ratio should by itself be able to promote protein-SSG formation in the absence of excessive oxidative stress (Figure 4). Thus, a high intracellular concentration of GSH under basal conditions should act as a protective mechanism against GSSG-induced protein-SSG formation. GSH can also induce protein-SSG formation, but only in the presence of oxidized cysteines (sulfenic acids), which might be a plausible mechanism under pro-oxidant apoptotic stimuli.102 Apoptosis is accompanied by increased protein-SSG formation.103 TNF-α-induced apoptosis is reported to be paralleled by increased protein-SSG formation, which is inhibited by overexpression of Bcl-2.103 Recently, FasL-induced apoptosis has also been reported to increase protein-SSG, which amplifies the apoptotic signaling cascade by glutathionylation of the Fas receptor.104 In contrast, caspases have been reported to be glutathionylated under basal conditions and to become deglutathionylationed upon the induction of apoptosis.105 Accordingly, GSSG-induced caspase 3 glutathionylation has been recently reported to inhibit its proteolytic activity.106

Molecular mechanisms involved in the regulation of apoptosis by GSH. GSH has been reported to regulate apoptosis by distinct mechanisms. (a) GSH might be a prerequisite for the generation of RS. In some cases RS induced by pro-oxidant apoptotic conditions lead directly to GSH depletion by its oxidation to GS• and further formation/accumulation of GSSG. (b) GSH depletion and the concomitant decrease in GSH/GSSG ratio might increase the availability of GSSG to exert its redox signaling, even in the absence of excessive RS formation and oxidative stress. (c) Alterations in GSH, GSSG, and RS concentrations and equilibrium have been shown to mediate oxidative post-translational modifications (OPMs) in proteins that may regulate the progression of apoptosis. The oxidated intermediate of GSH, GS•, might also be involved in the direct generation of OPMs. GSH and GSSG are known to promote reversible OPMs such as protein-SSG (glutathionylation) and protein-SNO (nitrosylation) formation that regulate different signaling proteins involved in apoptotic cell death. (d) Changes in GSH content also modulate glutathione S-transferases (GST) activity and its interaction with other apoptotic signaling proteins such as JNK
Glutathione content has been shown to regulate the activation of ionic conductances during apoptosis. Alterations in the intracellular ionic homeostasis of the cell have been clearly linked to the progression of apoptosis.107 Recent reports show that during apoptosis, cell shrinkage or apoptotic volume decrease, associated with the impairment in the Na+-K+-ATPase activity,108 and activation of K+ and Cl− conductances109, 110 are also regulated by alterations in GSH homeostasis. A wide variety of ion channels and transporters have been shown to be regulated by ROS.111 In addition, sulfhydryl groups within cytoplasmic cysteine domains of ion channels directly modulate channel activity.112 However, it is still unclear whether during apoptosis GSH depletion regulates ionic pathways indirectly through ROS or directly by protein glutathionylation.
Protein-SSG linkages are removed by changes in the intracellular GSH/GSSG balance and/or the activities of glutaredoxin (GRX) enzymes.113 GRXs are versatile oxidoreductases that use the reducing power of GSH to catalyze reversible protein glutathionylation (reduction of mixed disulfides). Inhibition of GRXs by cadmium induces apoptosis in the absence of any other stimuli, which is associated to increased protein-SSG formation.114 Human cells contain four GRXs. Of these, the cytosolic dithiol GRX1 and the mitochondrial monothiol GRX2 have been implicated in the regulation of apoptosis.115 GRX1 has been shown to protect against apoptosis induced by Fas activation by reducing Fas glutathionylation, which potentiates its aggregation and recruitment into lipid rafts.104 Overexpression of GRX1 also protects against doxorubicin-induced cell death.116 Conversely, GRX1 downregulation sensitizes lens epithelial cells to oxidative stress-induced apoptosis.117 Mitochondrial GRX2 is also involved in the regulation of apoptosis, and its downregulation sensitizes cells to apoptosis.118 GRX2 has been shown to protect against apoptosis by reducing Cyt c release and cardiolipin oxidation.119 Recently GRXs have been shown not only to deglutathionylate specific proteins but also to catalyze the glutathionylation of several proteins in the presence of GS• radicals. Hence, GRXs are capable of catalyzing both glutathionylation and deglutathionylation of proteins through distinct mechanisms.120 Thus, the role of both GRXs and protein-SSG formation needs to be simultaneously assessed to understand their specific involvement during apoptosis. Although GRXs have been shown to have a protective effect against apoptosis by decreasing protein-SSG formation, knockdown of GRX1 was recently reported to significantly inhibited TNF-α-induced cell death because of attenuated caspase 3 cleavage and increased glutathionylation.105 Therefore, tissue specific and spatio-temporal regulation of protein glutathionylation in apoptosis might be having a function in cell death progression.
Non-enzymatic reaction of nitric oxide with GSH results in the formation of nitrosoglutathione (GSNO), which can induce protein-SSG formation by reaction with protein-thyil radicals (protein-S•)121. GSNO has been shown to induce glutathionylation caspases.122 GSNO can also be directly conjugated inactivated by its direct conjugation to proteins by protein nitrosylation (protein-SNO). Nitrosylation has been suggested to act as an intermediate for glutathionylation because of the fact that protein-SNO can readily react with GSH leading to protein-SSG formation.123 As GSH can promote protein-SNO, through GSNO protein-SNO might be one of the mechanisms by which changes in GSH content regulate apoptosis. Protein nitrosylation has been clearly shown to regulate apoptosis.124 For example, caspases have been shown to be nitrosylated under basal conditions and to become denitrosylated on apoptosis.125
The role of GSH in other types of cell death pathways
Cell death is often classified by both biochemical and morphological criteria. According to the classification of cell death recommended by the Nomenclature Committee on Cell Death,126 three distinct routes of cellular catabolism can be defined according to morphological criteria, which are apoptosis, autophagy, and necrosis although there are numerous examples in which cell death displays mixed features. Autophagy is a major catabolic pathway by which eukaryotic cells degrade and recycle macromolecules and organelles. It has an essential role in differentiation and development, as well as in cellular response to stress. Autophagy can be activated during amino-acid deprivation and has been associated with neurodegenerative diseases, cancer, pathogen infections, and myopathies. Autophagy is initiated by the surrounding of cytoplasmic constituents by the crescent-shaped isolation membrane/phagophore, which forms a closed double-membrane structure, called autophagosome. Finally, the autophagosome fuses with a lysosome to become an autolysosome, and its content is degraded by acidic lysosomal hydrolases. Autophagic cell death is morphologically defined by massive autophagic vacuolization of the cytoplasm in the absence of chromatin condensation.126 Replenishment of intracellular GSH levels with NAC has been shown to abolish ROS-induced formation of autophagosomes and the consequent degradation of proteins during starvation-induced autophagy.127 Lipopolysaccharide-induced autophagy was also shown to be paralleled by ROS formation and GSH depletion, which was prevented by NAC.128 Treatment with gamma-glutamylcysteinyl ethyl ester, a precursor for de novo GSH formation, also prevents autophagy after traumatic brain injury.129
Necrotic cell death is characterized by a gain in cell volume, swelling of organelles, plasma membrane rupture, and subsequent loss of intracellular contents. It is now recognized that execution of necrotic cell death may be finely regulated by a series of signal transduction pathways and catabolic processes.126 Excessive GSH depletion and oxidative stress have also been reported to switch apoptotic to necrotic cell death.130 Earlier work has shown that GSH-depleting agents such as DEM at doses that deplete mitochondrial GSH levels will induce necrosis. However, modest doses of GSH-depleting agents resulting mainly in cytoplasmic GSH depletion and minimal change in cell viability sensitize hepatocytes to TNF-α-induced apoptosis.79 Ceramide has been implicated as a secondary messenger for TNF-α-induced cell necrosis and NAC or GSH-monoethylester have been shown to delay the onset of ceramide-induced necrosis.131
GSH, apoptosis, and disease progression
Alterations in intracellular GSH have been shown to occur during the progression of distinct pathological situations many of which are also related to alterations in apoptosis rates.4 We next summarize the evidence suggesting a role for GSH in the regulation of some of these human diseases.
Cancer
Glutathione and GSH-metabolism participate in both cancer prevention and progression. For example, under stress conditions, GSH scavenges harmful molecules such as electrophiles (by GSH conjugation), ROS, and RNS (by GSH antioxidant properties), preventing tumor initiation. Glutathione metabolism has been reported to participate in chemotherapy-induced tumor resistance to apoptotic cell death. Increased expression and polymorphisms of GST and GSH-transporters as well as high GSH concentration are common features of transformed cells that, in turn, are associated with high resistance to chemotherapy-induced apoptosis.132 High intracellular GSH content in tumor cells is a consequence of high γ-GCS expression and can promote tumor cell survival by inhibition of apoptosis. Modulation of intracellular GSH content has been largely studied for potential anti-cancer therapies. GSH depletors such as BSO, DEM, ethacrynic acid, and diamide have been shown to sensitize Bcl-2 overexpressing cell to undergo apoptosis.133 Another potential role for anti-neoplastic therapy is the modulation of GSH efflux by acting on its transporters or pumps. Recently, it has been shown that transformed cells are sensitized to cell death when intracellular GSH is depleted through stimulation of GSH efflux pumps,21, 134 whose expression varies in different cancer cell lines.135
Neurodegenerative Diseases
In the nervous system, GSH is necessary for the defense against oxidative stress and the progression of distinct pathologies including those of Parkinson's disease (PD) and Alzheimer's disease (AD). Neurodegenerative diseases are associated with selective neuron loss by apoptosis. Alterations in intracellular GSH homeostasis have been observed in different experimental models of neurodegenerative diseases.136 Recent reports have shown a direct link between a reduction in GSH content and apoptosis in neuronal cells associated with neurodegeneration.8, 137 Glutathione metabolism is reported to be altered in affected brain regions and peripheral cells from AD patients while its levels are depleted in experimental models of AD.138 AD is associated with the formation of amyloid plaques in the brains of AD patients. Amyloid β-peptide is the main constituent of amyloid plaques. Amyloid β-peptides induce GSH depletion in cultured neurons and its cytotoxicity and accumulation is stimulated by GSH loss.139 Cultured neurons from GSH peroxidase 1 GPX1(−/−) mice are more susceptible to cell death induced by amyloid β-peptides140 whereas overexpression of GPX1 in the PC12 pheochromocytoma cells or rat embryonic cultured cortical neurons renders these cells more resistant to these peptides.141 In addition, increased protein glutathionylation (protein-SSG) has been reported during AD, which is associated with increased oxidative stress.142 Accordingly, GRX1 overexpression protects against amyloid β-induced toxicity.143
PD progression is associated with a depletion of GSH levels and an increase in ROS formation in the substantia nigra. In fact GSH depletion has been proposed as one of the early biochemical events associated with neuronal apoptosis in PD.137 Decreases in total GSH concentrations in the substantia nigra have been observed in preclinical stages of PD, at a time at which other biochemical changes are not yet detectable.144 Thiol antioxidants, such as GSH and NAC, have been reported to prevent apoptosis induced by dopamine in neural cultures, which is proposed to mediate oxidative stress during PD by its auto-oxidation.145 Overexpression of GPX1 in neuroblastoma cells in vitro and in nigral dopaminergic neurons in vivo attenuates apoptosis in experimental models of PD.146 Glutathionylation of mitochondrial NADP-dependent isocitrate dehydrogenase has been observed in an animal model of PD147 and overexpression of GRX2 protected against apoptosis in distinct experimental models of PD.148
Liver Toxicity
Drugs, infections, and inflammation in the liver increase the generation of ROS and RNS with concomitant decreases in GSH levels causing a shift in the cellular redox status of hepatocytes. This shift alters several cell signaling pathways in hepatocytes leading to the pathogenesis of many liver diseases. Liver injury induced by acetaminophen involves severe GSH depletion of both cytosolic and mitochondrial pools, ROS generation, and cell death mediated by JNK and ASK-1 activation. It has been shown that Nrf2 is activated by acetaminophen, which in turn increases GSH synthesis in hepatocytes. In this way, NAC has been proven to be effective in treating patients with acetaminophen-induced liver injury that, conversely, is potentiated by GSH depletors.149 Alcohol-induced liver injury has often been associated with oxidative stress and decreases in hepatic antioxidant defense, especially GSH. GSH precursors have also been shown to protect against ethanol-induced toxicity in rat models. Chronic ethanol treatment is believed to deplete mitochondrial GSH by ethanol-induced changes in the inner membrane fluidity associated with cholesterol accumulation, which decreases GSH transporter activity. As liver mitochondria lack catalase, mitochondrial GSH and GSH peroxidases (GPX) are the major mechanisms for H2O2 in detoxification. Thus, ethanol toxicity is also paralleled by increased GSSG accumulation.150
Conclusions and Perspectives
Recent studies have shown an active role for GSH in redox regulation of different processes. GSH depletion during apoptosis has been observed to occur at early stages during the cell death program. GSH depletion was initially ascribed to its oxidation by RS generated during oxidative stress. However, it is now recognized that under more physiological stimulation of apoptosis, such as activation of death receptors, GSH depletion occurs as an active process involving its extrusion across the plasma membrane. This phenomenon has also been shown to precede oxidative stress generated by the accumulation of RS and to be necessary for the progression of apoptosis. Indeed, GSH depletion has been shown to induce or potentiate apoptosis, and excessive oxidative stress. Although the exact mechanisms involved in the regulation of apoptosis by GSH remain elusive, recent reports show that oxidative post-translational modifications in proteins regulated by GSH content such as glutathionylation (protein-SSG) and nitrosylation (protein-SNO) are important regulators of apoptosis. Thus, it is time to look beyond GSH as a simple antioxidant and define its precise role in programmed cell death signaling.
Abbreviations
- GSH:
-
reduced glutathione
- GSSG:
-
glutathione disulfide
- GS•:
-
thyil radical
- ROS:
-
reactive oxygen species
- RNS:
-
reactive nitrogen species
- RS:
-
reactive species
- γ-GCS:
-
γ-glutamylcysteine synthetase
- GPX:
-
glutathione peroxidase
- GR:
-
glutathione reductase
- GST:
-
glutathione S-transferase
- GSNO:
-
nitrosoglutathione
- G6PD:
-
glucose-6-phosphate dehydrogenase
- NAC:
-
N-acetyl-L-cysteine
- NO:
-
nitric oxide
- protein-SSG:
-
protein glutathionylation
- protein-SNO:
-
protein nitrosylation
References
Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14: 1237–1243.
Pervaiz S, Clement MV . A permissive apoptotic environment: function of a decrease in intracellular superoxide anion and cytosolic acidification. Biochem Biophys Res Commun 2002; 290: 1145–1150.
Circu ML, Aw TY . Glutathione and apoptosis. Free Radic Res 2008; 42: 689–706.
Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI . The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem 2007; 113: 234–258.
Forman HJ, Zhang H, Rinna A . Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 2008; 30: 1–12.
Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG . Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med 2004; 37: 1511–1526.
Armstrong JS, Whiteman M, Yang H, Jones DP, Sternberg Jr P . Cysteine starvation activates the redox-dependent mitochondrial permeability transition in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 2004; 45: 4183–4189.
Diaz-Hernandez JI, Almeida A, Delgado-Esteban M, Fernandez E, Bolanos JP . Knockdown of glutamate-cysteine ligase by small hairpin RNA reveals that both catalytic and modulatory subunits are essential for the survival of primary neurons. J Biol Chem 2005; 280: 38992–39001.
Botta D, Franklin CC, White CC, Krejsa CM, Dabrowski MJ, Pierce RH et al. Glutamate-cysteine ligase attenuates TNF-induced mitochondrial injury and apoptosis. Free Radic Biol Med 2004; 37: 632–642.
Fan Y, Wu D, Jin L, Yin Z . Human glutamylcysteine synthetase protects HEK293 cells against UV-induced cell death through inhibition of c-Jun NH2-terminal kinase. Cell Biol Int 2005; 29: 695–702.
Wild AC, Mulcahy RT . Regulation of gamma-glutamylcysteine synthetase subunit gene expression: Insights into transcriptional control of antioxidant defenses. Free Radic Res 2000; 32: 281–301.
Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM et al. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med 2004; 37: 1152–1159.
Wild AC, Moinova HR, Mulcahy RT . Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem 1999; 274: 33627–33636.
Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 2002; 365: 405–416.
Morito N, Yoh K, Itoh K, Hirayama A, Koyama A, Yamamoto M et al. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene 2003; 22: 9275–9281.
Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 2003; 23: 3394–3406.
Friesen C, Kiess Y, Debatin KM . 2004 A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ 2004; 11 (Suppl 1): S73–S85.
Cazanave S, Berson A, Haouzi D, Vadrot N, Fau D, Grodet A et al. High hepatic glutathione stores alleviate Fas-induced apoptosis in mice. J Hepatol 2006; 46: 858–868.
Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW et al. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ 2002; 9: 252–263.
Franco R, Panayiotidis MI, Cidlowski JA . Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem 2007; 282: 30452–30465.
Franco R, Cidlowski JA . SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J Biol Chem 2006; 281: 29542–29557.
Kirkland RA, Franklin JL . Evidence for redox regulation of cytochrome C release during programmed neuronal death: antioxidant effects of protein synthesis and caspase inhibition. J Neurosci 2001; 21: 1949–1963.
Wang X, Cederbaum AI . S-adenosyl-L-methionine attenuates hepatotoxicity induced by agonistic Jo2 Fas antibody following CYP2E1 induction in mice. J Pharmacol Exp Ther 2006; 317: 44–52.
Will Y, Kaetzel RS, Brown MK, Fraley TS, Reed DJ . In vivo reversal of glutathione deficiency and susceptibility to in vivo dexamethasone-induced apoptosis by N-acetylcysteine and L-2-oxothiazolidine-4-carboxylic acid, but not ascorbic acid, in thymocytes from gamma-glutamyltranspeptidase-deficient knockout mice. Arch Biochem Biophys 2002; 397: 399–406.
Lash LH . Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem Biol Interact 2006; 163: 54–67.
Garcia-Ruiz C, Fernandez-Checa JC . Mitochondrial glutathione: hepatocellular survival-death switch. J Gastroenterol Hepatol 2006; 21 (Suppl 3): S3–S6.
Marchetti P, Decaudin D, Macho A, Zamzami N, Hirsch T, Susin SA et al. Redox regulation of apoptosis: impact of thiol oxidation status on mitochondrial function. Eur J Immunol 1997; 27: 289–296.
Muyderman H, Wadey AL, Nilsson M, Sims NR . Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J Neurochem 2007; 102: 1369–1382.
Xu F, Putt DA, Matherly LH, Lash LH . Modulation of expression of rat mitochondrial 2-oxoglutarate carrier in NRK-52E cells alters mitochondrial transport and accumulation of glutathione and susceptibility to chemically induced apoptosis. J Pharmacol Exp Ther 2006; 316: 1175–1186.
Lash LH, Putt DA, Matherly LH . Protection of NRK-52E cells, a rat renal proximal tubular cell line, from chemical-induced apoptosis by overexpression of a mitochondrial glutathione transporter. J Pharmacol Exp Ther 2002; 303: 476–486.
Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A et al. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998; 115: 1541–1551.
Zhao P, Kalhorn TF, Slattery JT . Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology 2002; 36: 326–335.
Circu ML, Rodriguez C, Maloney R, Moyer MP, Aw TY . Contribution of mitochondrial GSH transport to matrix GSH status and colonic epithelial cell apoptosis. Free Radic Biol Med 2008; 44: 768–778.
Blair IA . Endogenous glutathione adducts. Curr Drug Metab 2006; 7: 853–872.
Valko M, Morris H, Cronin MT . Metals, toxicity and oxidative stress. Curr Med Chem 2005; 12: 1161–1208.
Hammond CL, Marchan R, Krance SM, Ballatori N . Glutathione export during apoptosis requires functional multidrug resistance-associated proteins. J Biol Chem 2007; 282: 14337–14347.
Cole SP, Deeley RG . Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci 2006; 27: 438–446.
Trompier D, Chang XB, Barattin R, du Moulinet D’Hardemare A, Di Pietro A, Baubichon-Cortay H . Verapamil and its derivative trigger apoptosis through glutathione extrusion by multidrug resistance protein MRP1. Cancer Res 2004; 64: 4950–4956.
Marchan R, Hammond CL, Ballatori N . Multidrug resistance-associated protein 1 as a major mediator of basal and apoptotic glutathione release. Biochim Biophys Acta 2008; 1778: 2413–2420.
Li L, Meier PJ, Ballatori N . Oatp2 mediates bidirectional organic solute transport: a role for intracellular glutathione. Mol Pharmacol 2000; 58: 335–340.
Mahagita C, Grassl SM, Piyachaturawat P, Ballatori N . Human organic anion transporter 1B1 (OATP1B1/OATP-C) and 1B3 (OATP1B3/OATP-8) function as bidirectional carriers and do not mediate GSH-bile acid co-transport. Am J Physiol Gastrointest Liver Physiol 2007; 293: G271–G278.
Franklin CC, Krejsa CM, Pierce RH, White CC, Fausto N, Kavanagh TJ . Caspase-3-dependent cleavage of the glutamate-L-cysteine ligase catalytic subunit during apoptotic cell death. Am J Pathol 2002; 160: 1887–1894.
Jungas T, Motta I, Duffieux F, Fanen P, Stoven V, Ojcius DM . Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J Biol Chem 2002; 277: 27912–27918.
Rana S, Dringen R . Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci Lett 2006; 415: 45–48.
Circu ML, Stringer S, Rhoads CA, Moyer MP, Aw TY . The role of GSH efflux in staurosporine-induced apoptosis in colonic epithelial cells. Biochem Pharmacol 2009; 77: 76–85.
Paolicchi A, Dominici S, Pieri L, Maellaro E, Pompella A . Glutathione catabolism as a signaling mechanism. Biochem Pharmacol 2002; 64: 1027–1035.
Karp DR, Shimooku K, Lipsky PE . Expression of gamma-glutamyl transpeptidase protects ramos B cells from oxidation-induced cell death. J Biol Chem 2001; 276: 3798–3804.
Armstrong JS, Jones DP . Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. Faseb J 2002; 16: 1263–1265.
Varghese J, Khandre NS, Sarin A . Caspase-3 activation is an early event and initiates apoptotic damage in a human leukemia cell line. Apoptosis 2003; 8: 363–370.
Sato T, Machida T, Takahashi S, Iyama S, Sato Y, Kuribayashi K et al. Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J Immunol 2004; 173: 285–296.
D’Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. Faseb J 2005; 19: 1504–1506.
Brown GC, Borutaite V . Regulation of apoptosis by the redox state of cytochrome c. Biochim Biophys Acta 2008; 1777: 877–881.
Vaughn AE, Deshmukh M . Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol 2008; 10: 1477–1483.
Martin SF, Sawai H, Villalba JM, Hannun YA . Redox regulation of neutral sphingomyelinase-1 activity in HEK293 cells through a GSH-dependent mechanism. Arch Biochem Biophys 2007; 459: 295–300.
Lou H, Kaplowitz N . Glutathione depletion down-regulates tumor necrosis factor alpha-induced NF-kappaB activity via IkappaB kinase-dependent and -independent mechanisms. J Biol Chem 2007; 282: 29470–29481.
Voehringer DW, Meyn RE . Redox aspects of Bcl-2 function. Antioxid Redox Signal 2000; 2: 537–550.
Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G et al. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem 1996; 67: 1259–1267.
Zimmermann AK, Loucks FA, Schroeder EK, Bouchard RJ, Tyler KL, Linseman DA . Glutathione binding to the Bcl-2 homology-3 domain groove: A molecular basis for Bcl-2 antioxidant function at mitochondria. J Biol Chem 2007; 282: 29296–29304.
Schor NF, Rudin CM, Hartman AR, Thompson CB, Tyurina YY, Kagan VE . Cell line dependence of Bcl-2-induced alteration of glutathione handling. Oncogene 2000; 19: 472–476.
Bojes HK, Datta K, Xu J, Chin A, Simonian P, Nunez G et al. Bcl-xL overexpression attenuates glutathione depletion in FL5.12 cells following interleukin-3 withdrawal. Biochem J 1997; 325 (Pt 2): 315–319.
Cerella C, Coppola S, D’Alessio M, De Nicola M, Magrini A, Bergamaschi A et al. Redox modulation of the apoptogenic activity of thapsigargin. Ann NY Acad Sci 2007; 1099: 469–472.
Chakravarthi S, Jessop CE, Bulleid NJ . The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep 2006; 7: 271–275.
Jessop CE, Bulleid NJ . Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J Biol Chem 2004; 279: 55341–55347.
Cullinan SB, Diehl JA . PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 2004; 279: 20108–20117.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ . Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21: 1249–1259.
Green RM, Graham M, O’Donovan MR, Chipman JK, Hodges NJ . Subcellular compartmentalization of glutathione: correlations with parameters of oxidative stress related to genotoxicity. Mutagenesis 2006; 21: 383–390.
Higuchi Y . Glutathione depletion-induced chromosomal DNA fragmentation associated with apoptosis and necrosis. J Cell Mol Med 2004; 8: 455–464.
Kamada K, Goto S, Okunaga T, Ihara Y, Tsuji K, Kawai Y et al. Nuclear glutathione S-transferase pi prevents apoptosis by reducing the oxidative stress-induced formation of exocyclic DNA products. Free Radic Biol Med 2004; 37: 1875–1884.
Hollins DL, Suliman HB, Piantadosi CA, Carraway MS . Glutathione regulates susceptibility to oxidant-induced mitochondrial DNA damage in human lymphocytes. Free Radic Biol Med 2006; 40: 1220–1226.
Jones DP . Redefining oxidative stress. Antioxid Redox Signal 2006; 8: 1865–1879.
Filomeni G, Aquilano K, Civitareale P, Rotilio G, Ciriolo MR . Activation of c-Jun-N-terminal kinase is required for apoptosis triggered by glutathione disulfide in neuroblastoma cells. Free Radic Biol Med 2005; 39: 345–354.
Pias EK, Aw TY . Early redox imbalance mediates hydroperoxide-induced apoptosis in mitotic competent undifferentiated PC-12 cells. Cell Death Differ 2002; 9: 1007–1016.
Ho HY, Cheng ML, Chiu DT . Glucose-6-phosphate dehydrogenase – from oxidative stress to cellular functions and degenerative diseases. Redox Rep 2007; 12: 109–118.
Ayene IS, Biaglow JE, Kachur AV, Stamato TD, Koch CJ . Mutation in G6PD gene leads to loss of cellular control of protein glutathionylation: Mechanism and implication. J Cell Biochem 2008; 103: 123–135.
Fico A, Paglialunga F, Cigliano L, Abrescia P, Verde P, Martini G et al. Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis. Cell Death Differ 2004; 11: 823–831.
Gendron MC, Schrantz N, Metivier D, Kroemer G, Maciorowska Z, Sureau F et al. Oxidation of pyridine nucleotides during Fas- and ceramide-induced apoptosis in Jurkat cells: Correlation with changes in mitochondria, glutathione depletion, intracellular acidification and caspase 3 activation. Biochem J 2001; 353: 357–367.
Banki K, Hutter E, Colombo E, Gonchoroff NJ, Perl A . Glutathione levels and sensitivity to apoptosis are regulated by changes in transaldolase expression. J Biol Chem 1996; 271: 32994–33001.
Delgado-Esteban M, Almeida A, Bolanos JP . D-Glucose prevents glutathione oxidation and mitochondrial damage after glutamate receptor stimulation in rat cortical primary neurons. J Neurochem 2000; 75: 1618–1624.
Han D, Hanawa N, Saberi B, Kaplowitz N . Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic Biol Med 2006; 41: 627–639.
Minich T, Riemer J, Schulz JB, Wielinga P, Wijnholds J, Dringen R . The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J Neurochem 2006; 97: 373–384.
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 2007; 9: 49–89.
Arthur JR . The glutathione peroxidases. Cell Mol Life Sci 2000; 57: 1825–1835.
Kayanoki Y, Fujii J, Islam KN, Suzuki K, Kawata S, Matsuzawa Y et al. The protective role of glutathione peroxidase in apoptosis induced by reactive oxygen species. J Biochem 1996; 119: 817–822.
Yan W, Chen X . GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J Biol Chem 2006; 281: 7856–7862.
Crack PJ, Taylor JM, Flentjar NJ, de Haan J, Hertzog P, Iannello RC et al. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J Neurochem 2001; 78: 1389–1399.
Gouaze V, Mirault ME, Carpentier S, Salvayre R, Levade T, Andrieu-Abadie N . Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol Pharmacol 2001; 60: 488–496.
Faucher K, Rabinovitch-Chable H, Cook-Moreau J, Barriere G, Sturtz F, Rigaud M . Overexpression of human GPX1 modifies Bax to Bcl-2 apoptotic ratio in human endothelial cells. Mol Cell Biochem 2005; 277: 81–87.
Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts 2nd LJ et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem 2004; 279: 55137–55146.
Ran Q, Gu M, Van Remmen H, Strong R, Roberts JL, Richardson A. . Glutathione peroxidase 4 protects cortical neurons from oxidative injury and amyloid toxicity. J Neurosci Res 2006; 84: 202–208.
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 2008; 8: 237–248.
Savaskan NE, Borchert A, Brauer AU, Kuhn H . Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: Specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic Biol Med 2007; 43: 191–201.
Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y . Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J 2000; 351: 183–193.
Shen HM, Pervaiz S . TNF receptor superfamily-induced cell death: Redox-dependent execution. Faseb J 2006; 20: 1589–1598.
Devadas S, Hinshaw JA, Zaritskaya L, Williams MS . Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med 2003; 35: 648–661.
Gouaze V, Andrieu-Abadie N, Cuvillier O, Malagarie-Cazenave S, Frisach MF, Mirault ME et al. Glutathione peroxidase-1 protects from CD95-induced apoptosis. J Biol Chem 2002; 277: 42867–42874.
Bajt ML, Ho YS, Vonderfecht SL, Jaeschke H . Reactive oxygen as modulator of TNF and fas receptor-mediated apoptosis in vivo: studies with glutathione peroxidase-deficient mice. Antioxid Redox Signal 2002; 4: 733–740.
Han YH, Kim SZ, Kim SH, Park WH . Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the changes of intracellular GSH levels rather than ROS levels. Lung Cancer 2007; 59: 301–314.
Jacobson MD, Raff MC . Programmed cell death and Bcl-2 protection in very low oxygen. Nature 1995; 374: 814–816.
Jiang S, Cai J, Wallace DC, Jones DP . Cytochrome c-mediated apoptosis in cells lacking mitochondrial DNA. Signaling pathway involving release and caspase 3 activation is conserved. J Biol Chem 1999; 274: 29905–29911.
Han YH, Kim SH, Kim SZ, Park WH . Apoptosis in arsenic trioxide-treated Calu-6 lung cells is correlated with the depletion of GSH levels rather than the changes of ROS levels. J Cell Biochem 2008; 104: 862–878.
Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 2008; 45: 1–17.
Dalle-Donne I, Giustarini D, Colombo R, Milzani A, Rossi R . S-glutathionylation in human platelets by a thiol-disulfide exchange-independent mechanism. Free Radic Biol Med 2005; 38: 1501–1510.
Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T . Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 2000; 39: 11121–11128.
Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS et al. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol 2009; 184: 241–252.
Pan S, Berk BC . Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res 2007; 100: 213–219.
Huang Z, Pinto JT, Deng H, Richie Jr JP . Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem Pharmacol 2008; 75: 2234–2244.
Franco R, Bortner CD, Cidlowski JA . Potential roles of electrogenic ion transport and plasma membrane depolarization in apoptosis. J Membr Biol 2006; 209: 43–58.
Yin W, Cheng W, Shen W, Shu L, Zhao J, Zhang J et al. Impairment of Na(+),K(+)-ATPase in CD95(APO-1)-induced human T-cell leukemia cell apoptosis mediated by glutathione depletion and generation of hydrogen peroxide. Leukemia 2007; 21: 1669–1678.
Shimizu T, Numata T, Okada Y . A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(-) channel. Proc Natl Acad Sci USA 2004; 101: 6770–6773.
Franco R, DeHaven WI, Sifre M, Bortner CD, Cidlowski JA . Glutathione depletion and disruption of intracellular ionic homeostasis regulate lymphoid cell apoptosis. J Biol Chem 2007; 283: 36071–36087.
Kourie JI . Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol 1998; 275: C1–24.
Cai S, Sauve R . Effects of thiol-modifying agents on a K(Ca2+) channel of intermediate conductance in bovine aortic endothelial cells. J Membr Biol 1997; 158: 147–158.
Gallogly MM, Mieyal JJ . Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol 2007; 7: 381–391.
Chrestensen CA, Starke DW, Mieyal JJ . Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem 2000; 275: 26556–26565.
Lillig CH, Berndt C, Holmgren A . Glutaredoxin systems. Biochim Biophys Acta 2008; 1780: 1304–1317.
Meyer EB, Wells WW . Thioltransferase overexpression increases resistance of MCF-7 cells to adriamycin. Free Radic Biol Med 1999; 26: 770–776.
Lofgren S, Fernando MR, Xing KY, Wang Y, Kuszynski CA, Ho YS et al. Effect of thioltransferase (glutaredoxin) deletion on cellular sensitivity to oxidative stress and cell proliferation in lens epithelial cells of thioltransferase knockout mouse. Invest Ophthalmol Vis Sci 2008; 49: 4497–4505.
Lillig CH, Lonn ME, Enoksson M, Fernandes AP, Holmgren A . Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc Natl Acad Sci USA 2004; 101: 13227–13232.
Enoksson M, Fernandes AP, Prast S, Lillig CH, Holmgren A, Orrenius S . Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem Biophys Res Commun 2005; 327: 774–779.
Dalle-Donne I, Milzani A, Gagliano N, Colombo R, Giustarini D, Rossi R . Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signal 2008; 10: 445–473.
West MB, Hill BG, Xuan YT, Bhatnagar A . Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. Faseb J 2006; 20: 1715–1717.
Klatt P, Pineda Molina E, Perez-Sala D, Lamas S . Novel application of S-nitrosoglutathione-Sepharose to identify proteins that are potential targets for S-nitrosoglutathione-induced mixed-disulphide formation. Biochem J 2000; 349: 567–578.
Martinez-Ruiz A, Lamas S . Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc Res 2007; 75: 220–228.
Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS . Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005; 6: 150–166.
Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K et al. S-Nitrosylation of mitochondrial caspases. J Cell Biol 2001; 154: 1111–1116.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z . Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26: 1749–1760.
Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol 2009; 296: H470–H479.
Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab 2008; 28: 540–550.
Troyano A, Sancho P, Fernandez C, de Blas E, Bernardi P, Aller P . The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ 2003; 10: 889–898.
Davis MA, Flaws JA, Young M, Collins K, Colburn NH . Effect of ceramide on intracellular glutathione determines apoptotic or necrotic cell death of JB6 tumor cells. Toxicol Sci 2000; 53: 48–55.
Estrela JM, Ortega A, Obrador E . Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci 2006; 43: 143–181.
Voehringer DW . BCL-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic Biol Med 1999; 27: 945–950.
Benlloch M, Ortega A, Ferrer P, Segarra R, Obrador E, Asensi M et al. Acceleration of glutathione efflux and inhibition of gamma-glutamyltranspeptidase sensitize metastatic B16 melanoma cells to endothelium-induced cytotoxicity. J Biol Chem 2005; 280: 6950–6959.
Perez-Tomas R . Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem 2006; 13: 1859–1876.
Dringen R, Hirrlinger J . Glutathione pathways in the brain. Biol Chem 2003; 384: 505–516.
Merad-Boudia M, Nicole A, Santiard-Baron D, Saille C, Ceballos-Picot I . Mitochondrial impairment as an early event in the process of apoptosis induced by glutathione depletion in neuronal cells: relevance to Parkinson's disease. Biochem Pharmacol 1998; 56: 645–655.
Aksenov MY, Markesbery WR . Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer's disease. Neurosci Lett 2001; 302: 141–145.
Woltjer RL, Nghiem W, Maezawa I, Milatovic D, Vaisar T, Montine KS et al. Role of glutathione in intracellular amyloid-alpha precursor protein/carboxy-terminal fragment aggregation and associated cytotoxicity. J Neurochem 2005; 93: 1047–1056.
Crack PJ, Cimdins K, Ali U, Hertzog PJ, Iannello RC . Lack of glutathione peroxidase-1 exacerbates a beta-mediated neurotoxicity in cortical neurons. J Neural Transm 2006; 113: 645–657.
Barkats M, Millecamps S, Abrioux P, Geoffroy MC, Mallet J . Overexpression of glutathione peroxidase increases the resistance of neuronal cells to A beta-mediated neurotoxicity. J Neurochem 2000; 75: 1438–1446.
Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM et al. An increase in S-glutathionylated proteins in the Alzheimer's disease inferior parietal lobule, a proteomics approach. J Neurosci Res 2007; 85: 1506–1514.
Akterin S, Cowburn RF, Miranda-Vizuete A, Jimenez A, Bogdanovic N, Winblad B et al. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ 2006; 13: 1454–1465.
Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol 1994; 35: 38–44.
Offen D, Ziv I, Sternin H, Melamed E, Hochman A . Prevention of dopamine-induced cell death by thiol antioxidants: possible implications for treatment of Parkinson's disease. Exp Neurol 1996; 141: 32–39.
Ridet JL, Bensadoun JC, Deglon N, Aebischer P, Zurn AD . Lentivirus-mediated expression of glutathione peroxidase: neuroprotection in murine models of Parkinson's disease. Neurobiol Dis 2006; 21: 29–34.
Kil IS, Park JW . Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem 2005; 280: 10846–10854.
Daily D, Vlamis-Gardikas A, Offen D, Mittelman L, Melamed E, Holmgren A et al. Glutaredoxin protects cerebellar granule neurons from dopamine-induced apoptosis by activating NF-kappa B via Ref-1. J Biol Chem 2001; 276: 1335–1344.
Jaeschke H, Bajt ML . Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci 2006; 89: 31–41.
Iimuro Y, Bradford BU, Yamashina S, Rusyn I, Nakagami M, Enomoto N et al. The glutathione precursor L-2-oxothiazolidine-4-carboxylic acid protects against liver injury due to chronic enteral ethanol exposure in the rat. Hepatology 2000; 31: 391–398.
Acknowledgements
This work was supported by the Intramural Research Program of the NIH/National Institute of Environmental Health Sciences 1Z01ES090079. We acknowledge Dr. Carl D Bortner, Dr. Robert H Oakley, and Dr. John B Pritchard critical comments of this manuscript. Owing to space considerations we apologize for not citing many important contributions to this field. The supplemental data contain a list of these important works. Color figures are included in the HTML version of this manuscript
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by A Finazzi-Agro
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Franco, R., Cidlowski, J. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ 16, 1303–1314 (2009). https://doi.org/10.1038/cdd.2009.107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.107
Keywords
This article is cited by
-
Radio-resistance of hypoxic tumors: exploring the effects of oxygen and X-ray radiation on non-small lung cancer cell lines
Radiation Oncology (2023)
-
Blockage of glycolysis by targeting PFKFB3 suppresses the development of infantile hemangioma
Journal of Translational Medicine (2023)
-
Mediation and longitudinal analysis to interpret the association between clozapine pharmacokinetics, pharmacogenomics, and absolute neutrophil count
Schizophrenia (2023)
-
Oral GSH Exerts a Therapeutic Effect on Experimental Salmonella Meningitis by Protecting BBB Integrity and Inhibiting Salmonella-induced Apoptosis
Journal of Neuroimmune Pharmacology (2023)
-
Urechistachykinin I induced ferroptosis by accumulating reactive oxygen species in Vibrio vulnificus
Applied Microbiology and Biotechnology (2023)
