
Abstract
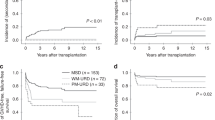
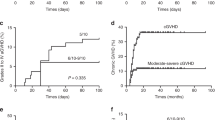
From January 1991 to March 2007, 61 children and adolescent with acquired severe aplastic anemia received BMT in our institutions. We retrospectively compared the outcome of 30 cases of matched-sibling donor BMT (MSD-BMT) and 31 cases of unrelated donor BMT (URD-BMT). We observed one graft failure among MSD-BMT recipients and three graft failures among URD-BMT recipients, respectively. No patients in the MSD-BMT group developed grades II–IV acute GVHD compared with 11 of 30 patients (37%) in the URD-BMT group (P<0.001). One of 30 MSD-BMT recipients (3%) developed chronic GVHD compared with 8 of 30 URD-BMT recipients (27%) (P=0.013). The incidence of EBV and CMV reactivation was 11 of 20 URD-BMT recipients and 23 of 30, respectively. One patient in the URD-BMT group died of a motor accident 5.5 years after BMT. Ten-year OS was 100% in MSD-BMT recipients and 93.8% (95% CI, 81.9–100%) in URD-BMT recipients, respectively (P=0.252). Ten-year failure-free survival was 96.7% (95% CI, 90.2–100%) in the MSD-BMT group and 84.7% (95% CI, 70.2–99.2%) in the URD-BMT group, respectively (P=0.161).
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Frickhofen N, Kaltwasser JP, Schrezenmeier H, Raghavachar A, Vogt HG, Herrmann F et al. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. The German Aplastic Anemia Study Group. N Engl J Med 1991; 324: 1297–1304.
Bacigalupo A, Broccia G, Corda G, Arcese W, Carotenuto M, Gallamini A et al. Antilymphocyte globulin, cyclosporin, and granulocyte colony-stimulating factor in patients with acquired severe aplastic anemia (SAA): a pilot study of the EBMT SAA Working Party. Blood 1995; 85: 1348–1353.
Margolis D, Camitta B, Pietryga D, Keever-Taylor C, Baxter-Lowe LA, Pierce K et al. Unrelated donor bone marrow transplantation to treat severe aplastic anaemia in children and young adults. Br J Haematol 1996; 94: 65–72.
Deeg HJ, Seidel K, Casper J, Anasetti C, Davies S, Gajeweski JL et al. Marrow transplantation from unrelated donors for patients with severe aplastic anemia who have failed immunosuppressive therapy. Biol Blood Marrow Transplant 1999; 5: 243–252.
Horowitz MM . Current status of allogeneic bone marrow transplantation in acquire aplastic anemia. Semin Hematol 2000; 37: 30–42.
Passweg JR, Perez WS, Eapen M, Camitta BM, Gluckman E, Hinterberger W et al. Bone marrow transplants from mismatched related and unrelated donors for severe aplastic anemia. Bone Marrow Transplant 2006; 37: 641–649.
Maury S, Balere-Appert ML, Chir Z, Boiron JM, Galambrun C, Yakouben K et al. Unrelated stem cell transplantation for severe acquired aplastic anemia: improved outcome in the era of high-resolution HLA matching between donor and recipient. Haematologica 2007; 92: 589–596.
Kennedy-Nasser AA, Leung KS, Mahajan A, Weiss HL, Arce JA, Gottschalk S et al. Comparable outcomes of matched-related and alternative donor stem cell transplantation for pediatric severe aplastic anemia. Biol Blood Marrow Transplant 2006; 12: 1277–1284.
Camitta BM, Thomas ED, Nathan DG, Santos G, Gordon-Smith EC, Gale RP et al. Severe aplastic anemia: a prospective study of the effect of early marrowtransplantation on acute mortality. Blood 1976; 48: 63–70.
Miyamura K, Kojima S, Takeyama K, Matsushita T, Fukuda M, Horibe K et al. Use of cyclophosphamide and total lymphoid irradiation combined with cyclosporine in bone marrow transplantation for transfused severe aplastic anemia. Bone Marrow Transplant 1988; 3: 457–461.
Azuma E, Kojima S, Kato K, Matsuyama T, Yamada Y, Kondo N et al. Conditioning with cyclophosphamide/antithymocyte globulin for allogeneic bone marrow transplantation from HLA-matched siblings in children with severe aplastic anemia. Bone Marrow Transplant 1997; 19: 1085–1087.
Kojima S, Inaba J, Yoshimi A, Takahashi Y, Watanabe N, Kudo K et al. Unrelated donor marrow transplantation in children with severe aplastic anaemia using cyclophosphamide, anti-thymocyte globulin and total body irradiation. Br J Haematol 2001; 114: 706–711.
Storb R, Deeg HJ, Farewell V, Doney K, Appelbaum F, Beatty P et al. Marrow transplantation for severe aplastic anemia: methotrexate alone compared with a combination of methotrexate and cyclosporine for prevention of acute graft-versus-host disease. Blood 1986; 68: 119–125.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HLA-matched sibling donors. Transplant 1974; 18: 295–304.
Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE et al. Chronic graft-versus-host syndrome in man: a long-term clinicopathologic study of 20 Seattle patients. Am J Med 1980; 69: 204–217.
Hoshino Y, Kimua H, Tanaka N, Tsuge I, Kudo K, Horibe K et al. Prospective monitoring of the Epstein-Barr virus DNA by a real-time quantitative polymerase chain reaction after allogenic stem cell transplantation. Br J Haematol 2001; 115: 105–111.
Kojima S, Matsuyama T, Kato S, Kigasawa H, Kobayashi R, Kikuta A et al. Outcome of 154 patients with severe aplastic anemia who received transplants from unrelated donors: the Japan Marrow Donor Program. Blood 2002; 100: 799–805.
Viollier R, Socie G, Tichelli A, Bacigalupo A, Korthof ET, Marsh J et al. Recent improvement in outcome of unrelated donor transplantation for aplastic anemia. Bone Marrow Transplant 2008; 41: 45–50.
Vassiliou GS, Webb DK, Pamphilon D, Knapper S, Veys PA . Improved outcome of alternative donor bone marrow transplantation in children with severe aplastic anaemia using a conditioning regimen containing low-dose total body irradiation, cyclophosphamide and Campath. Br J Haematol 2001; 114: 701–705.
Brunstein CG, Weisdorf DJ, DeFor T, Barker JN, Tolar J, van Burik JA et al. Marked increased risk of Epstein-Barr virus-related complications with the addition of antithymocyte globulin to a nonmyeloablative conditioning prior to unrelated umbilical cord blood transplantation. Blood 2006; 108: 2874–2880.
Buyck HC, Ball S, Junagade P, Marsh J, Chakrabarti S . Prior immunosuppressive therapy with antithymocyte globulin increases the risk of EBV-related lymphoproliferative disorder following allo-SCT for acquired aplastic anaemia. Bone Marrow Transplant 2009; 43: 813–816.
Armand P, Antin JH . Allogeneic stem cell transplantation for aplastic anemia. Biol Blood Marrow Transplant 2007; 13: 505–516.
Socié G, Henry-Amar M, Cosset JM, Devergie A, Girinsky T, Gluckman E . Increased incidence of solid malignant tumors after bone marrow transplantation for severe aplastic anemia. Blood 1991; 78: 277–279.
Kobayashi R, Yabe H, Hara J, Morimoto A, Tsuchida M, Mugishima H et al. Preceding immunosuppressive therapy with antithymocyte globulin and cyclosporin increases the incidence of graft rejection in children with aplastic anaemia who underwent allogeneic bone marrow transplantation from HLA-identical siblings. Br J Haematol 2006; 135: 693–696.
Bacigalupo A, Locatelli F, Lanino E, Marsh J, Socié G, Maury S et al. Fludarabine, cyclophosphamide and anti-thymocyte globulin for alternative donor transplants in acquired severe aplastic anemia: a report from the EBMT-SAA Working Party. Bone Marrow Transplant 2005; 36: 947–950.
Deeg HJ, O’Donnell M, Tolar J, Agarwal R, Harris RE, Feig SA et al. Optimization of conditioning for marrow transplantation from unrelated donors for patients with aplastic anemia after failure of immunosuppressive therapy. Blood 2006; 108: 1485–1491.
Lee JH, Choi SJ, Lee JH, Lee YS, Seol M, Ryu SG et al. Non-total body irradiation containing preparative regimen in alternative donor bone marrow transplantation for severe aplastic anemia. Bone Marrow Transplant 2005; 35: 755–761.
Siegal D, Xu W, Sutherland R, Kamel-Reid S, Kuruvilla J, Lipton JH et al. Graft-versus-host disease following marrow transplantation for aplastic anemia: different impact of two GVHD prevention strategies. Bone Marrow Transplant 2008; 42: 51–56.
Gupta V, Ball SE, Sage D, Ortin M, Freires M, Gordon-Smith EC et al. Marrow transplants from matched unrelated donors for aplastic anaemia using alemtuzumab, fludarabine and cyclophosphamide based conditioning. Bone Marrow Transplant 2005; 35: 467–471.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yagasaki, H., Takahashi, Y., Hama, A. et al. Comparison of matched-sibling donor BMT and unrelated donor BMT in children and adolescent with acquired severe aplastic anemia. Bone Marrow Transplant 45, 1508–1513 (2010). https://doi.org/10.1038/bmt.2009.378
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bmt.2009.378
Keywords
This article is cited by
-
Recent advances in the diagnosis and treatment of pediatric acquired aplastic anemia
International Journal of Hematology (2024)
-
Comparison of HLA-matched sibling and unrelated donor transplantation in adult patients with acquired severe aplastic anemia
Bone Marrow Transplantation (2020)
-
Conditioning regimen for allogeneic bone marrow transplantation in children with acquired bone marrow failure: fludarabine/melphalan vs. fludarabine/cyclophosphamide
Bone Marrow Transplantation (2020)
-
Comparison of efficacy and health-related quality of life of first-line haploidentical hematopoietic stem cell transplantation with unrelated cord blood infusion and first-line immunosuppressive therapy for acquired severe aplastic anemia
Leukemia (2020)
-
Who is the best haploidentical donor for acquired severe aplastic anemia? Experience from a multicenter study
Journal of Hematology & Oncology (2019)
