
Abstract
Background:
The Notch pathway is frequently activated in cancer. Pathway inhibition by γ-secretase inhibitors has been shown to be effective in pre-clinical models of pancreatic cancer, in combination with gemcitabine.
Methods:
A multi-centre, non-randomised Bayesian adaptive design study of MK-0752, administered per os weekly, in combination with gemcitabine administered intravenously on days 1, 8 and 15 (28 day cycle) at 800 or 1000 mg m-2, was performed to determine the safety of combination treatment and the recommended phase 2 dose (RP2D). Secondary and tertiary objectives included tumour response, plasma and tumour MK-0752 concentration, and inhibition of the Notch pathway in hair follicles and tumour.
Results:
Overall, 44 eligible patients (performance status 0 or 1 with adequate organ function) received gemcitabine and MK-0752 as first or second line treatment for pancreatic cancer. RP2Ds of MK-0752 and gemcitabine as single agents could be combined safely. The Bayesian algorithm allowed further dose escalation, but pharmacokinetic analysis showed no increase in MK-0752 AUC (area under the curve) beyond 1800 mg once weekly. Tumour response evaluation was available in 19 patients; 13 achieved stable disease and 1 patient achieved a confirmed partial response.
Conclusions:
Gemcitabine and a γ-secretase inhibitor (MK-0752) can be combined at their full, single-agent RP2Ds.
Similar content being viewed by others


Main
Pancreatic cancer is one of the most lethal human cancers because of late presentation, early metastases and resistance of tumour cells to most conventional treatments. Until recently, the standard treatment in the metastatic setting remained gemcitabine. Other approaches now considered include the FOLFIRINOX regime and the combination of gemcitabine and abraxane (Conroy et al, 2011; Von Hoff et al, 2013). As the majority of patients with pancreatic cancer develop advanced metastatic disease, and chemotherapeutics have limited impact on survival, improvements in systemic-targeted therapies are desperately needed.
Studies of the molecular genetics of human pancreatic ductal adenocarcinoma (PDAC) have identified a number of common genetic alterations (Jones et al, 2008); however, targeted therapies have not been shown to be beneficial. In addition, advances have been made in the understanding of pancreatic developmental biology and the identification of developmental signalling pathways that are important in PDAC, including the Notch pathway (Miyamoto et al, 2003; Cook et al, 2012). The Notch pathway is involved in the development, differentiation, growth and fate of multiple different cell types (Bray, 2006), and has a fairly limited set of known target genes, including transcriptional regulators Hes (hairy/enhancer of split) and Hey (subfamily of Hes) (Borggrefe and Oswald, 2009). Notch signalling is relatively inactive in the normal adult pancreas; however, it becomes activated in damaged and dysplastic pancreatic tissue (Miyamoto et al, 2003). In humans, PDAC activation of the Notch pathway has been shown to correlate with neurovascular metastasis (Buchler et al, 2005), and over-expression of Notch3 has been linked to a more aggressive phenotype and resistance to chemotherapy (Doucas et al, 2008; Yao and Qian, 2009). This perhaps indicates that Notch not only has a role in development of PDAC, but also in its maintenance and progression.
Most Notch ligands and receptors are upregulated in PDAC therefore pan-Notch inhibition is likely to be more useful than inhibiting a single component of the pathway. Pan-Notch inhibition is possible pharmacologically with γ-secretase inhibitors (GSIs). γ-secretase is the crucial proteolytic activity that releases the Notch intracellular domain and is therefore a key player in the canonical Notch signalling pathway. GSIs prevent cleavage of the intracellular Notch domains by blocking the proteolytic function of presenilin enzymes, thereby keeping Notch in an intact, latent form. GSIs have been shown to be effective in genetically engineered mouse models of PDAC, alone and in combination with gemcitabine (Plentz et al, 2009; Cook et al, 2012).
MK-0752 is a highly potent and specific GSI (Krop et al, 2012). MK-0752 appears to be well tolerated as a single-agent and toxicities appear to be schedule dependent (Krop et al, 2012). A preference for a once weekly dosing schedule is dictated by tolerability, pharmacodynamic (PD) activity and pharmacokinetic (PK) results (Krop et al, 2012). Observed PK and PD data from phase I studies of MK-0752 alone suggested no significant increase in drug exposure between 1800 mg and 3200 mg, and the maximum tolerated dose (MTD) on the weekly schedule was defined as 3200 mg. The recommended dose for further phase II evaluation (RP2D) of MK-0752 in single-agent studies is 1800 mg; however, the combination of MK-0752 and gemcitabine has not previously been reported. Gemcitabine (2′,2′,-difluoro-2′,-deoxycytidine) is a nucleoside analogue of deoxycytidine that interferes with DNA synthesis through inhibition of ribonucleotide reductase and competition with dCTP for incorporation into DNA. It has broad applications as a cytotoxic chemotherapeutic agent in multiple solid tumour types, and is used as a standard treatment in pancreatic cancer (Burris et al, 1997).
The primary objective of this trial was to recommend a dose for further phase II evaluation for the combination of MK-0752 and gemcitabine; and determine the safety and tolerability of combination treatment. Secondary objectives included evaluation of tumour response, measuring time to disease progression, overall survival at 6 months and 1 year, and percentage change in CA19-9 levels. Tertiary objectives included investigations into the PK profile of MK-0752 in plasma, the feasibility of measuring MK-0752 levels in tumour and assessment of PD markers of target inhibition by MK-0752.
Patients and methods
Patient eligibility
Patients with histologically or cytologically proven stage III (inoperable) and IV PDAC were eligible for the trial. Previous chemotherapy for advanced disease was permitted. If gemcitabine treatment had been given previously, the patient must have tolerated a dose of at least 800 mg m-2. Patients were required to have radiologically assessable disease and tissue that was accessible to biopsy. Other inclusion criteria included a life expectancy of at least 12 weeks, World Health Organisation (WHO) performance status of 0 or 1 and the following haematological and biochemical parameters; haemoglobin ⩾9 g dl-1; absolute neutrophil count ⩾1.5 × 109 l-1; platelet count ⩾100 × 109 l-1; serum bilirubin ⩽1.5 × upper limit of normal (ULN); alanine amino-transferase (ALT) ⩽2.5 × ULN (unless raised due to liver metastases in which case up to 5 × ULN was permissible); prothrombin time (PT) ⩽1.5 × ULN and calculated creatinine clearance ⩾50 ml min-1. All patients gave written informed consent in accordance with the institutional guidelines before study treatment.
Study design
This was a multi-centre, non-randomised Bayesian adaptive phase I study of MK-0752, administered per os (p.o.) weekly in combination with gemcitabine administered intravenously (IV) on days 1, 8 and 15 (800 or 1000 mg m-2) with MK-0752 administered alone on day 22 (28 day cycle) (https://clinicaltrials.gov/ct2/show/NCT01098344). Cycle 1 had a duration of 42 days (treatment with MK-0752 alone administered on day -14 and day -7); subsequent cycles were 28 days in duration. The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines and approved by relevant regulatory and independent ethics committee (NRES Committee East of England—Cambridge East Ref: 10/H0304/3). Six UK sites participated.
The Bayesian bivariate dose escalation procedure was conducted through the direction of a computer programme operated by the Medical and Pharmaceutical (MPS) Research Unit at Lancaster University (Whitehead et al, 2012). The starting dose of the phase I dose escalation stage of the study was based on the results of previous and ongoing phase I trials of MK-0752 in patients with solid tumours. The decision to dose escalate and the number of patients to be treated at any given dose level (beyond one patient) was made by the Investigators after review of all available toxicity data and output from the Bayesian algorithm (Supplementary Figure 1).
The DLT and MTD were defined using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) Version 4.02. The MTD was defined as the dose most likely to be associated with a risk of a DLT, during the first cycle of a patient’s treatment, of 20%. A DLT was defined as almost certainly or probably drug-related and occurring in the first cycle of treatment, including grade 4 neutropenia for ⩾5 days duration; febrile neutropenia; grade 4 thrombocytopenia for ⩾5 days; grade 3 or 4 toxicity to organs other than the bone marrow, including grade 3 and 4 biochemical adverse events (AEs) as DLTs but excluding grade 3 nausea, vomiting or diarrhoea in patients who have not received optimal treatment. The phase I dose escalation part of the study was considered to be complete either when the same treatment combination had been recommended and administered nine times (to different patients), or when the safety and clinical profile indicate that the RP2D of MK-0752 in single-agent studies given in combination with either 800 mg m-2 or 1000 mg m-2 gemcitabine should become the recommended dose combination for phase 2.
Patient evaluation
All patients receiving at least one administration of MK-0752 were evaluable for toxicity. Patients who did not complete the first cycle of treatment for reasons other than DLTs were not evaluable for the determination of the MTD and were replaced. All patients who met eligibility criteria, received at least one cycle of treatment with MK-0752 alone followed by MK-0752 in combination with gemcitabine, were assessed for efficacy. To be evaluable for response, patients were to have received at least two cycles of study medication, had a baseline assessment of disease and at least one repeat disease assessment.
History, physical examination (symptom directed), WHO performance status and weight were performed pre-dose on days -14, -7, 1, 8, 15 and 22 of cycles 1 and 2, then day 1 of each cycle thereafter. AEs were graded according to NCI CTCAE Version 4.02. CA19-9 was measured before treatment on day -14 and then on day 1 of each cycle. Radiological assessments (CT, MRI, X-ray) were repeated every 8 weeks and tumour response was assessed based on the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 (Eisenhauer et al, 2009). Time to progressive disease (as per RECIST v1.1) and survival at 6 months and 1 year were calculated from cycle 1, day -14 when MK-0752 was administered for the first time.
Dose modifications and duration of treatment
Dose and dose level refer to both MK-0752 and gemcitabine unless otherwise stated. Grade 1 AEs required no dose modification. At first occurrence of a grade 2–4 non-haematological or gastrointestinal-related AEs, the dose of both MK-0752 and gemcitabine were withheld until the AE grade returned to a grade 1. With grade 2 haematological AEs, treatment could be continued at the same dose level unless the platelet count was <75 × 109l-1. If initially a grade 3 or 4 AE, the dose was then reduced by one dose level (for both MK-0752 and gemcitabine) or discontinued at the discretion of the investigator. The gemcitabine dose level was not reduced below 600 mg m-2. If a patient experienced several AEs, then the recommended dose adjustment to reduce the dose to the lowest level was used and the schedule maintained. MK-0752 was reduced first unless either stated otherwise or the AE was considered related (almost certainly, probably or possibly) to gemcitabine only. If the starting dose was not tolerated then the patient could continue to receive gemcitabine as a single agent. Patients requiring a delay of >2 weeks were removed from study.
Combination treatment continued for six cycles unless the patient was withdrawn, there was evidence of disease progression or the patient was experiencing unacceptable toxicity. If a patient was benefiting from treatment (i.e., has stable or responding disease as measured by RECIST 1.1) and the benefit – risk balance was considered acceptable then further treatment could be given following approval from the Sponsor for up to six additional cycles. Patients were followed up for 28 days after the last administration of MK-0752 or gemcitabine. All patients were followed up for disease progression and survival every two months, continued for 12 months following cycle 1, day 1. Beyond 12 months, patients were followed up for progression free and overall survival data collection (during their standard clinic visits) and the period for collecting this data continued for 12 months from cycle 1 day 1 of the final patient recruited to the study.
PK analysis
Plasma samples for analysis of MK-0752 levels were collected at the following time points in cycle 1: day −14: immediately prior to MK-0752 dose; day -7: immediately prior to MK-0752 dose and then at 1, 4, 8 and 24 h post-dose; post day −7: approximately at the same time as the second tumour biopsy sample (within 24–48 h of day -7); day 1: immediately prior to MK-0752 dose and then at 1, 4, 8 and 24 h post-dose. Samples were collected within 10% of the proposed time following MK-0752 dosing. Tumour biopsy was collected at baseline and within 72 h post day -7 dose of MK-0752. Tumour and plasma concentrations of MK-0752 were evaluated by Merck, Kenilworth, NJ, USA. MK-0752 was isolated from blood or tumour by protein precipitation and analysed by liquid chromatography/tandem mass spectrometry (LC–MS/MS). Non-compartmental PK analysis was performed using Phoenix v6.0 software on non-rounded bioanalytical data. The PK parameters determined for MK-0752, alone and in combination with gemcitabine, include the maximum observed plasma concentration (Cmax), time to reach Cmax (Tmax), and the area under the plasma concentration time curve (AUC). The same calculations were used to measure plasma and tissue concentrations.
PD analysis
Tumour biopsy samples were used to undertake transcriptional profiling to assess Notch pathway activity, and immunohistochemical (IHC) analysis of certain Notch target genes. The timing of tissue collection is described in the PK methods section. The tumour tissue, where accessible by endoscopic ultrasound (EUS) or CT-guided biopsy, was obtained by the same technique on each occasion with three to five passes made at each attempt. The following samples were collected from each patient; (a) specimen for formalin fixed paraffin embedded (FFPE) slides; (b) specimen for RNA analysis stored in RNAeasy (Qiagen, Manchester, UK); c. specimen for PK analysis (methods described in the PK section). These were collected following standard internal hospital procedures. The FFPE samples for IHC were processed and analysed at the CRUK Cambridge Institute following internal laboratory procedures. Notch pathway inhibition was evaluated in tumour tissue by assessing IHC for certain target genes (Hes and Hey family). The Hes1 antibody (clone NM1; MBLI; 1:250 dilution), and Hey1 antibody (ab22614; Abcam; 1:50 dilution) were used. To ensure uniformity of staining, all slides were stained in a single batch. Scoring of the immunohistochemistry was carried out blinded. A grade of 0-3 was given to each sample depending on the frequency and intensity of staining; 0: no staining; 1: low frequency and/or intensity of staining; 2: frequent and moderate intensity or moderate frequency and high intensity; 3: very frequent and high intensity. In the case of Hes1, nuclear staining in tumour cells was scored. Nuclear and cytoplasmic Hey1 was assessed in tumour and stromal cells. IHC assay scoring was analysed using the statistical software package GraphPad Prism (San Diego, CA, USA) version five.
All other PD tumour samples and hair follicles were shipped directly from the CRUK Cambridge Institute to LabCorp (formerly Covance Genomics Lab) Seattle, WA, USA for RNA extraction and Affy microarray processing. Plucked hair follicles were obtained from patients participating at baseline and 6–8 h after the MK-0752 administration on day -7, always taken from the patients head. The published refined signature in hair follicles exhibited a strong linear correlation with plasma MK-0752, confirming its utility as a surrogate PD biomarker. (Krop et al, 2012). Hair follicles are extracted using the Promega SV total RNA isolation kit. The tumour samples were extracted using homogenisation in RLT buffer, Trizol extraction and final isolation using the Promega SV96 isolation kit. All RNA samples were treated with DNAse as part of the process. Fifty ng of total RNA was amplified using the NuGEN Ovation Whole Blood Solution protocol (NuGEN, San Carlos, CA, USA) as described by the manufacturer. Amplified biotin-labelled material was hybridised to custom designed Affymetrix microarrays, one sample per array. Hybridisation, washing and scanning were completed as recommended by the manufacturer LabCorp Clinical Trials in Seattle.
Results
Patient characteristics
A total of 44 eligible patients were enroled in this study between 26-May-2010 and 10-Oct-2014, of whom 42 received treatment and were therefore evaluable for safety analysis. Baseline characteristics are described in Table 1. The majority of patients had stage IV pancreatic adenocarcinoma (93%) and 30% had received prior chemotherapy.
Dose escalation and MTD
During dose escalation, where safety constraints permitted, patients were commenced on study in pairs as per the Bayesian algorithm, described previously (Whitehead et al, 2012) (Supplementary Figures 1 and 2). At each stage of dose escalation, the Bayesian algorithm recommended MK-0752 doses for use with 800 and 1000 mg m-2 of gemcitabine, which were thought most likely to be associated with a DLT risk of 0.2 on the basis of data available from patients evaluable for determination of the MTD. None of the patients in dose levels 1–3, a and b arms or dose level 4b, experienced a dose-limiting toxicity (DLT). The RP2D of both single agents was shown to be safe in combination (MK-0752 1800 mg p.o. once weekly and gemcitabine 1000 mg m-2 (i.v. days 1, 8 and 15 or a 28 day cycle). The Bayesian algorithm would permit further dose escalation, but accruing PK data (see below) confirmed previous findings that dose escalation beyond the RP2D of MK-0752 did not lead to higher plasma AUCs (Krop et al, 2012). Two dose-limiting toxicities were observed: grade 3 hypokalaemia was observed at MK-0752 2400 mg/gemcitabine 800 mg m-2 (dose level 5b) in one patient and grade 3 fatigue was observed in 1 patients at MK-0752 1800 mg/gemcitabine 1000 mg m-2 (dose level 4a) (Supplementary Figure 2). Of the 42 patients who received treatment, 31 patients (74%) had at least one dose delay and/or dose modification (mostly reductions and delays and missed doses). Three patients (7%) required at least one dose reduction of MK-0752 and 4 patients, at least one dose reduction of gemcitabine (9%). One patient required dose reduction of both drugs. Dose escalation continued to dose level 5b (MK-0752 2400 mg/gemcitabine 800 mg m-2). Dose level 6 was not explored. The MTD of both drugs combined was recommended to be the same as the RP2D of both drugs as single agents (MK-0752 1800 mg and gemcitabine 1000 mg m-2) on the basis of clinical and safety profiles. At the point the study closed, according to the Bayesian procedure the dose combination with the highest probability of having a toxicity risk of 0.2 was the RP2D of both drugs as single agents.
Safety and tolerability
The incidences of grade 3 and 4 AEs deemed to be related to either study drug are listed by dose level (Table 2). Gastrointestinal-related AEs were common although primarily grade 1 or 2 (nausea 23 pts (55%); vomiting 23 pts (55%); diarrhoea 20 pts (48%)). Other AEs affecting over 20% of patients included; thrombocytopenia (40.5%), anaemia (41%), anorexia (33%), fatigue (31%), neutropenia (29%) and transaminitis. Seven patients died during the active treatment period of the study, that is, within 28 days of their last IMP administration. Three deaths were related solely to progressive disease and 1 patient died due to progressive disease associated with an intracranial haemorrhage and disseminated intravascular coagulation (DIC). The remaining 3 patients died due to a thromboembolic event leading to cardiac arrest, sepsis and aspiration pneumonia. These deaths were not considered related to study treatment.
Pharmacokinetics
Data from 40 patients were available for plasma PK analysis. Plasma MK-0752 exposure (AUCo-t) and Cmax did not appear to be affected by the addition of gemcitabine, as parameters at day 1 (MK-0752+gemcitabine) are comparable with parameters at day -7 (MK-0752 alone) (Figure 1A and B). Comparing MK-0752 exposure across the dose groups, there was no increment in exposure beyond the RP2D single-agent dose (1800 mg weekly). Summary data for the RP2D of MK-0752 (1800 mg) are shown in Figure 1C.

Plasma and tumour MK-0752 PK analysis. (A) Plot of area under the curve (AUC0-t) vs MK-0752 dose (plasma). (B) Plot of Cmax (ng ml-1) vs MK-0752 dose (plasma). (C) Statistical summary of PK parameters at RP2D of MK-0752 (1800 mg). (D) MK-0752 levels in tumour tissue (ng g-1 tissue) plotted against dose of MK-0752. AUC0-t=area under the time:plasma concentration curve from time 0 to last measurable time point; Cmax=maximum concentration; CV=coefficient of variation as a percentage; ng=nanograms; PK=pharmcokinetics; RE=relative error; SD=standard deviation; Tmax= time of maximum concentration.
Eighteen paired patient biopsies were available for baseline and day -7, tumour PK analysis. MK-0752 was detected in day -7 tumour samples, with concentrations ranging from 480 to 30 600 ng g-1 (Figure 1D). The data are limited, but there appeared to be no increment in the tumour MK-0752 concentration between the 1500 and 2100 mg doses.
Pharmacodynamics
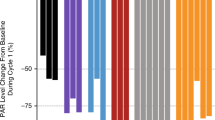
The ‘Notch signature score’ was analysed in hair follicles from 37 patients. Significant inhibition of Notch signalling post MK-0752 treatment, as evidenced by negative signature scores, was observed in 25/29 pts (Figure 2). There was no relationship between MK-0752 dose and the fold-change in Notch score, although number of samples was limited.

Notch signature score in hair follicle analysis. (A) Post-dose induced changes in NOTCH signature score in hair follicles of individual patients. Each bar represents individual patient treated with MK-0752 and the height of the bar (Y-axis) is equal to the difference between NOTCH signature score evaluated on patient sample post-treatment minus the baseline (pre-treatment) value of NOTCH signature score measured on a pre-treatment sample from the same patient. Patients are sorted by the post-dose induced changes in NOTCH signature score. Negative values on Y-axis indicate that the signature score was downregulated post-treatment and positive values indicate that signature score increased post-treatment in given patient. Downregulation of NOTCH signalling as captured by NOTCH signature score was observed in 25 out of 29 patients. (B) Scatter plot illustrating dose dependency of post-dose induced changes in NOTCH signature score for individual patients treated with MK-0752 (Y-axis) vs. MK-0752 dose (in mg) on X-axis. Each dot represents a patient. Data are shown for 18 patients with both pre- and post-dose profiling data available in hair follicle samples.
Forty-six pre- and post-treatment samples from 25 patients were received for IHC analysis. The biopsy samples were a mixture of Endoscopic Ultrasound-guided Fine Needle Aspirate (EUS FNA) from primary tumours and image-guided core biopsies from metastatic deposits. These biopsy samples were too small to be able to examine the endothelium, only the tumour cells were analysed. The 46 samples were comprised of 20 pairs of matched pre- and post-treatment biopsies. Five patients only had pre-treatment biopsies, and did not contribute to the comparison. Nine pairs could not be evaluated due to insufficient tissue. Hes1 and Hey1 target genes were not highly expressed in all baseline tumour biopsy samples, as had been expected. Of the 20 evaluated blocks for Hes1 staining, 4 were scored as Grade 0, indicating a lack of nuclear Hes1 staining. Seven samples were scored as Grade 1 and 8 as Grade 2. Only 1 of the evaluable blocks was scored as Grade 3 (Supplementary Figure 1).
CA19-9 levels and percentage changes were measured during the study, however patient numbers were small and it is not possible to draw any conclusions based on the limited number of results obtained.
Efficacy
In this study, 26 out of 44 evaluable patients were assessed for efficacy, as they received at least 1 complete cycle of treatment with MK-0752 alone followed by MK-0752 in combination with gemcitabine. The commonest reason for becoming non-evaluable during cycle 1 was symptomatic deterioration unrelated to study medication. Of the 26 patients evaluable patients, 19 received at least 2 cycles of treatment, provided baseline evaluations of disease and at least 2 repeat sets of disease assessments (patients were required to have at least one scan following their baseline scan to be evaluable, plus a second scan to confirm the findings of the first where there was a response (CR or PR)), so only these patients were evaluable for tumour response (RECIST 1.1). The median time to progressive disease was 169 days. Thirteen patients achieved stable disease, but only one patient achieved a confirmed partial response. This patient completed eight cycles of treatment. Nine patients were treated at the RP2D, and were evaluable for determination of the MTD. Of those, only 3 patients survived >6 months and 2 of these patients survived ⩾1 year. All patients in the RP2D population had stage IV pancreatic cancer at baseline. In terms of overall efficacy, the median overall survival time was 246 days in the response population (n=19), including patients who had been treated at all dose levels.
Discussion
The Notch signalling pathway is known to be an important target in pancreatic adenocarcinoma (PDAC), with components of the pathway found to be upregulated in various different cell types (Miyamoto et al, 2003; Mullendore et al, 2009; Cook et al, 2012). There are multiple published pre-clinical studies in PDAC, using various Notch pathway inhibitors alone and in combination, with encouraging results (Plentz et al, 2009; Yao and Qian, 2010; Cook et al, 2012; Yabuuchi et al, 2013; Palagani et al, 2014,). The most common therapeutic used in these studies have been GSIs. With these supportive pre-clinical findings, a phase I trial of MK-0752 in combination with gemcitabine in PDAC patients was undertaken. At the time of opening, it was the first trial to examine a GSI in patients with PDAC.
We observed that gemcitabine and MK-0752, can be combined at full, single-agent RP2Ds (MK-0752 1800 mg p.o. weekly and gemcitabine 1000 mg m-2 i.v. days 1, 8 and 15 of a 28 day cycle) and the regimen is well tolerated. The adaptive Bayesian algorithm used in the trial would have allowed further dose escalation beyond the single-agent RP2Ds, however the PK analysis revealed a plateau in plasma MK-0752 at the 1800 mg dose. As no severe adverse events were experienced the trial could have been more adaptive in its design by dropping the 800 mg gemcitabine cohort to potentially allow more rapid dose escalation using all the available data from all patients in each dose decision. The regimen was well tolerated although grade 1 and 2 gastrointestinal-related adverse events were common. No unexpected adverse events were identified. The occurrence of gastrointestinal, haematological and biochemical changes in liver function tests (transaminitis) are all recognised adverse events associated with MK-0752 and gemcitabine, when administered as single agents. Fatigue and thrombocytopenia were also commonly seen.
Of the 19 patients that had response assessments, 1 patient had a confirmed partial response and 13 patients stable disease. Three of 9 patients in the RP2D population survived for longer than 6 months, and 2 out of 9 remained alive at 1 year. This outcome may have been predicted following gemcitabine alone. The PK analysis revealed a plateau in plasma PK at the 1800 mg MK-0752 level. These results are similar to those previously reported in a study utilising MK-0752 as a single agent (Krop et al, 2012). The PK profile of MK-0752 was unaffected by gemcitabine. Importantly it was feasible to measure the concentration of MK-0752 in a limited number of tissue samples, proving that this drug does enter tumour tissue.
Multiple PD endpoints were examined both as surrogates for tumour activity (hair follicle gene expression array) and in the tumour (Hes1 and Hey1 target genes IHC and gene expression array). Paired biopsies were undertaken in over 50% of the patients on study, but the quality of these biopsies was variable. Pancreatic biopsies are notoriously difficult to obtain, and even in metastatic samples we did not always obtain the amount of tumour tissue required for informative PD assay investigations. In the samples with adequate amounts of tissue for analysis the expression of the Notch target genes we analysed was inconsistent with pre-clinical results (Cook et al, 2012). Several screening samples were graded 0–1, which confounds the assessment of the impact of therapy. Although two of the samples at the higher doses appeared to have less nuclear Hes1 staining post-treatment, the small numbers make it impossible to draw any conclusions as they were not consistently downregulated post-treatment.
Significant inhibition of Notch signalling post MK-0752 treatment was observed using the gene expression array from hair follicles. In summary, Notch pathway modulation was not identified in the tumour samples that were analysed, in spite of there being evidence of relevant exposure to MK-0752 being present in the samples. However, the unexpectedly low level expression of Hes1 and Hey1 is likely to have confounded these analyses.
GSIs have been the most broadly developed Notch pathway inhibitor (De Kloe and De Strooper, 2014). However, due to their lack of specificity, gastrointestinal side effects, and disappointing response rates further development may be challenging. RO4929097 is a GSI that has been used in single agent and combination early phase studies, including one in combination with gemcitabine in solid tumours, and one as GSI monotherapy in PDAC. (De Jesus-Acosta et al, 2014; Richter et al, 2014). Many trials involving this agent have been published, however due to lack of clinical activity in the phase II setting development of the compound has been discontinued (Strosberg et al, 2012; Tolcher et al, 2012; Diaz-Padilla et al, 2013, 2015; Lee et al, 2015). There are multiple other GSIs in clinical development both alone and in combination in various different tumour types (Gavai et al, 2015; Messersmith et al, 2015; Pant et al, 2016). None are yet to make it to development in the phase III setting in cancer.
GSIs are known to catalyse the proteolysis of many other substrates, therefore it is possible anti-tumour effects from GSIs could be attributed to one of these rather than solely inhibition of Notch signalling (Groth et al, 2010). Therefore, although the Notch pathway remains an interesting and relevant target in pancreatic cancer, newer active targeted agents directed against the pathway are needed. Given the expression of multiple different ligands and receptors from the Notch pathway in pancreatic cancer, and the role it has in the tumour cells and the microenvironment, it would be challenging to decide exactly which components of the pathway to target if not using a pan-Notch inhibitor. Also given the fact that not all Notch components are equivalent (Notch1 vs Notch3 receptors and Jagged 1 vs Dll4 ligands) it is also possible that specifically targeting individual or subsets of components is needed to elicit the desired effect.
Although we were able to administer MK-0752 and gemcitabine in combination, at their single agents RP2Ds, the activity seen, albeit in a small number of patients, was similar to that which would have been anticipated with gemcitabine alone. As this study commenced in 2010, alternative first line treatments for metastatic PDAC have been identified. These include FOLFIRINOX and nab-paclitaxel with gemcitabine (Conroy et al, 2011; Von Hoff et al, 2013). Therefore, we did not feel that the gemcitabine and MK-0752 combination explored in our study, merits further evaluation. However, Notch pathway inhibitors and chemotherapy combinations are being evaluated and data from our trial and the literature would suggest that the chemotherapy dosages will not need to be attenuated, with the addition of a Notch pathway inhibitor (Richter et al, 2014).
In summary, gemcitabine and MK-0752, can be combined at full, single-agent RP2Ds and the regimen was well tolerated. Some clinical activity was seen although the data were similar to what would have been expected with single-agent gemcitabine. Our data suggest that chemotherapy doses may not need to be attenuated in combinations including Notch pathway inhibitors.
References
Borggrefe T, Oswald F (2009) The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci 66: 1631–1646.
Bray SJ (2006) Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7: 678–689.
Buchler P, Gazdhar A, Schubert M, Giese N, Reber HA, Hines OJ, Giese T, Ceyhan GO, Muller M, Buchler MW, Friess H (2005) The Notch signaling pathway is related to neurovascular progression of pancreatic cancer. Ann Surg 242: 791–800.
Burris HA 3RD, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15: 2403–2413.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, De La Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer PRODIGE Intergroup (2011) Folfirinox versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825.
Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, Rao SS, Demuth T, Howat WJ, Jodrell DI, Tuveson DA (2012) Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med 209: 437–444.
DE Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, Blatchford PJ, Quackenbush K, Messersmith W (2014) A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs 32: 739–745.
De Kloe GE, De Strooper B (2014) Small molecules that inhibit Notch signaling. Methods Mol Biol 1187: 311–322.
Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, Zhang T, Kamel-Reid S, Ivy SP, Hotte SJ, Razak AA, Chen EX, Brana I, Wizemann M, Wang L, Siu LL, Bedard PL (2013) A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs 31: 1182–1191.
Diaz-Padilla I, Wilson MK, Clarke BA, Hirte HW, Welch SA, Mackay HJ, Biagi JJ, Reedijk M, Weberpals JI, Fleming GF, Wang L, LIU G, Zhou C, Blattler C, IVY SP, Oza AM (2015) A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: a study of the Princess Margaret, Chicago and California phase II consortia. Gynecol Oncol 137: 216–222.
Doucas H, Mann CD, Sutton CD, Garcea G, Neal CP, Berry DP, Manson MM (2008) Expression of nuclear Notch3 in pancreatic adenocarcinomas is associated with adverse clinical features, and correlates with the expression of STAT3 and phosphorylated Akt. J Surg Oncol 97: 63–68.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247.
Gavai AV, Quesnelle C, Norris D, Han WC, Gill P, Shan W, Balog A, Chen K, Tebben A, Rampulla R, Wu DR, Zhang Y, Mathur A, White R, Rose A, Wang H, Yang Z, Ranasinghe A, D'Arienzo C, Guarino V, Xiao L, Su C, Everlof G, Arora V, Shen DR, Cvijic ME, Menard K, Wen ML, Meredith J, Trainor G, Lombardo LJ, Olson R, Baran PS, Hunt JT, Vite GD, Fischer BS, Westhouse RA, Lee FY (2015) Discovery of clinical candidate BMS-906024: a potent Pan-notch inhibitor for the treatment of leukemia and solid tumors. ACS Med Chem Lett 6: 523–527.
Groth C, Alvord WG, Quinones OA, Fortini ME (2010) Pharmacological analysis of Drosophila melanogaster gamma-secretase with respect to differential proteolysis of Notch and APP. Mol Pharmacol 77: 567–574.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, FU B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, KinzlerK W (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806.
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, Lorusso P (2012) Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 30: 2307–2313.
Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, Othus M, Ribas A, Sondak VK, Gajewski TF, Margolin KA (2015) Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer 121: 432–440.
Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA, Mclachlan KR, Kern KA, Lorusso PM (2015) A phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-03084014. Clin Cancer Res 21: 60–67.
Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, ISO T, Meszoely IM, Wolfe MS, Hruban RH, Ball DW, Schmid RM, Leach SD (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3: 565–576.
Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, FAN X, Henderson CM, Matsui W, Eberhart CG, Maitra A, Feldmann G (2009) Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res 15: 2291–2301.
Palagani V, Bozko P, EL Khatib M, Belahmer H, Giese N, Sipos B, Malek NP, Plentz RR (2014) Combined inhibition of Notch and JAK/STAT is superior to monotherapies and impairs pancreatic cancer progression. Carcinogenesis 35: 859–866.
Pant S, Jones SF, Kurkjian CD, Infante JR, Moore KN, Burris HA, Mcmeekin DS, Benhadji KA, Patel BK, Frenzel MJ, Kursar JD, Zamek-Gliszczynski MJ, Yuen ES, Chan EM, Bendell JC (2016) A first-in-human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. Eur J Cancer 56: 1–9.
Plentz R, Park JS, Rhim AD, Abravanel D, Hezel AF, Sharma SV, Gurumurthy S, Deshpande V, Kenific C, Settleman J, Majumder PK, Stanger BZ, Bardeesy N (2009) Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 136: 1741–9 e6.
Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, Stathis A, Hirte HW, Razak AR, Reedijk M, Chen Z, Cohen B, Zhang WJ, Wang L, IVY SP, Moore MJ, Oza AM, Siu LL, Mcwhirter E (2014) A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs 32: 243–249.
Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, Almhanna K, Kim R, Valone T, Jump H, Sullivan D (2012) A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer 48: 997–1003.
Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari A, Shapiro GI, Boylan JF, Xu ZX, Wang K, Koehler A, Song J, Middleton SA, Deutsch J, Demario M, Kurzrock R, Wheler JJ (2012) Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol 30: 2348–2353.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369: 1691–1703.
Whitehead J, Thygesen H, Jaki T, Davies S, Halford S, Turner H, Cook N, Jodrell D (2012) A novel phase I/IIa design for early phase oncology studies and its application in the evaluation of MK-0752 in pancreatic cancer. Stat Med 31: 1931–1943.
Yabuuchi S, Pai SG, Campbell NR, De Wilde RF, DE Oliveira E, Korangath P, Streppel MM, Rasheed ZA, Hidalgo M, Maitra A, Rajeshkumar NV (2013) Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett 335: 41–51.
Yao J, Qian C (2009) Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol 27 (3): 1017–1022.
Yao J, Qian C (2010) Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol 27: 1017–1022.
Acknowledgements
We would like to acknowledge the support of Merck, Kenilworth, NJ, USA. This research was supported by the NIHR Leeds CRF Cambridge NIHR Biomedical Research Centre and the ECMC Network (funded by Cancer Research UK, the Department of Health, England and the Chief Scientist’s Office, Scotland) for this clinical trial. This study was funded and sponsored by the CRUK Centre of Drug Development with additional support for research and PK/PD analysis from Merck, Kenilworth, NJ, USA.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
DJ received support from Merck for meeting attendance and honoraria. Merck also contributed to some of the clinical trial costs associated with this study. DJ is a group leader in the Cancer Research UK funded CRUK Cambridge Institute, receiving core support from that Institute. MN is a full time employee of Merck, Kenilworth, NJ, USA. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Cook, N., Basu, B., Smith, DM. et al. A phase I trial of the γ-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer 118, 793–801 (2018). https://doi.org/10.1038/bjc.2017.495
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.495
Keywords
This article is cited by
-
The Notch signaling pathway: a potential target for cancer immunotherapy
Journal of Hematology & Oncology (2023)
-
Pancreatic cancer stemness: dynamic status in malignant progression
Journal of Experimental & Clinical Cancer Research (2023)
-
The strategies to cure cancer patients by eradicating cancer stem-like cells
Molecular Cancer (2023)
-
A microprotein N1DARP encoded by LINC00261 promotes Notch1 intracellular domain (N1ICD) degradation via disrupting USP10-N1ICD interaction to inhibit chemoresistance in Notch1-hyperactivated pancreatic cancer
Cell Discovery (2023)
-
The m6A reader IGF2BP3 preserves NOTCH3 mRNA stability to sustain Notch3 signaling and promote tumor metastasis in nasopharyngeal carcinoma
Oncogene (2023)

{kind=link}