
Abstract
Background:
Metastatic colorectal cancer (mCRC) patients with mutant KRAS or NRAS are ineligible for anti-epidermal growth factor receptor (anti-EGFR) therapy, as RAS mutations activate downstream pathways independently of EGFR and induce primary resistance. However, even among RAS wild-type (WT) patients, only a fraction responds to anti-EGFR therapy, suggesting that other mechanisms of resistance exist. We hypothesise that different (epi)genetic alterations can lead to primary anti-EGFR resistance and that the crucial end point is the activation of protein signalling pathways.
Methods:
We analysed the expression and activation of proteins involved in cell signalling, using reverse phase protein arrays, on a multicentre French cohort of RAS WT mCRC treated with anti-EGFR treatment.
Results:
We identify activated EGFR and HER3 as protein biomarkers predictive for better overall survival. Active EGFR signalling and downstream PI3K, but not MAPK, pathway activation are associated with response to anti-EGFR treatment. Left-sided mCRC displays active ErbB2/3 and Wnt pathways and a better response to anti-EGFR therapy compared to right-sided mCRC.
Conclusions:
We identify active EGFR and PI3K signalling as a key factor for response to anti-EGFR treatment in mCRC and highlight the importance of developing these biomarkers in clinical practice for the selection of RAS WT mCRC patients that would benefit from anti-EGFR treatment.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths, with 1.4 million new cases worldwide in 2012. The prognosis of CRC is mainly related to the presence of metastasis: around 25% of patients present with metastasis upon diagnosis and around 50% of patients that are treated for localised CRC will develop metastases during the course of disease. Despite the advances in early diagnosis and treatment achieved in the past 20 years, prognosis of metastatic CRC (mCRC) remains relatively poor, with a 5-year relative survival rate of about 12% (American Cancer Society).
Epidermal growth factor receptor (EGFR or ERBB1) is a cell membrane receptor that belongs to the family of receptor tyrosine kinases (Arteaga and Engelman, 2014). Upon binding of various ligands such as EGF, the receptor is activated and induces the activation of downstream signalling pathways, including PI3K/AKT, MEK/ERK, Jak/Stat and JUNK pathways, which contribute to tumorigenesis. Overexpression or activating mutations of EGFR occur in many cancer types, among which CRC. The development of monoclonal antibodies directed against EGFR, such as cetuximab and panitumumab, has significantly improved CRC outcome, both in the context of chemoresistant tumours (Amado et al, 2008; Karapetis et al, 2008) and as a first-line treatment (Bokemeyer et al, 2009; Van Cutsem et al, 2009). However, only patients having a tumour without mutations in KRAS and NRAS can benefit from anti-EGFR therapy (Douillard et al, 2013). Indeed, KRAS and NRAS operate downstream of EGFR in the RAS/MAPK signalling pathway and their mutation activates the pathway independently of EGFR status. KRAS and NRAS mutations are frequent, occurring in around 50% of CRCs, and their sequencing is therefore mandatory before administration of anti-EGFR treatment.
However, even among the patients with wild-type (WT) KRAS and NRAS, only 20–30% respond to the anti-EGFR treatment monotherapy (Price et al, 2016) and 65–70% to anti-EGFR combined with chemotherapy (Heinemann et al, 2016), suggesting that other molecular mechanisms of resistance exist. The identification of additional markers of resistance would allow to better select those patients that could benefit from anti-EGFR therapy and avoid inefficient and potentially toxic treatment of the other patients. CRC cell lines, xenografts and, less frequently, patient samples have been used to tackle this question. Multiple studies have shown that activation of the signalling pathways downstream of EGFR, induced by genetic alterations such as PTEN deletions, PIK3CA mutations or MET activation, constitute an important mechanism of primary and acquired resistance towards anti-EGFR (Bardelli and Siena, 2010; Troiani et al, 2013; Bajpe et al, 2014; Luraghi et al, 2014; Song et al, 2014; Van Emburgh et al, 2014). HER2 amplification or mutation has also been associated with anti-EGFR resistance in CRC xenografts (Bertotti et al, 2011; Yonesaka et al, 2011; Bertotti et al, 2015). Furthermore, amplifications or mutations of FGFR1, PDGFRA and MAP2K1 have been described (Bertotti et al, 2015), as well as the deregulation of several microRNAs. Yet, besides RAS, no other marker of resistance has been validated so far for clinical practice. In addition, recent data suggest that right-sided mCRC is more resistant to anti-EGFR therapy than left-sided mCRC (Moretto et al, 2016; Tejpar et al, 2016; Holch et al, 2017), but the biology underlying this difference remains elusive.
We hypothesise that many different genetic or epigenetic alterations can be involved in anti-EGFR resistance and that the crucial end point resides in the activation of downstream signalling pathways. The activation of these pathways would thus be a better and more universal predictor of resistance than each genetic alteration separately. However, large-scale protein data on CRC patient samples with clinical follow-up are currently missing. For this reason, we here decided to analyse the expression and the activation of a large panel of proteins involved in cell signalling pathways, using reverse phase protein arrays (RPPA) on a multicentre French cohort of RAS WT mCRCs, both left- and right-sided, treated with anti-EGFR treatment.
Materials and methods
Patient samples
Patients (n=53) with metastatic chemoresistant CRC were treated with anti-EGFR therapy (cetuximab or panitumumab), alone or in association with chemotherapy, at Institut Curie (Paris, France), CHU of Toulouse (France) or CHRU of Tours (France). Patients could be included in the study if tumour response to anti-EGFR could be specifically evaluated, that is, patients treated with a combination of anti-EGFR and chemotherapy who previously progressed on the same chemotherapy component (including those who had progressed on an oxaliplatin-based adjuvant chemotherapy then on a first-line irinotecan-based chemotherapy and those who had progressed on a first-line FOLFIRINOX tritherapy), or patients treated with a monotherapy of anti-EGFR or patients who initially progressed on a first-line combination of anti-EGFR and chemotherapy. According to French regulations, patients were informed of research performed with the biological specimens obtained during their treatment and did not express opposition. This retrospective study was reviewed and approved by the Ethics Committee of the Institut Curie. Time from sample resection to sample freezing was <30 min in most cases, and always <60 min. Samples were stored in secured −80 °C freezers. For this retrospective study, four 50 μm-thick frozen tissue sections of the primary tumour, obtained before administration of anti-EGFR treatment and containing at least 50% of tumour cells, were sent to the RPPA platform of Curie. Sequencing of KRAS, NRAS, BRAF and PIK3CA was performed independently on 3 of the 28 regional molecular genetics platforms constituting the national network of public laboratories dedicated to molecular oncology tests in France that has been certified by the French National Cancer Institute (INCa). According to the INCa quality assurance programme, these platforms have used one of the recommended sequencing techniques with detection sensitivity between 5 and 10% of mutated cells (allelic hybridisation using HRM followed by Sanger sequencing or by pyrosequencing). From the 53 samples, 7 were removed due to low RPPA signals, probably reflecting protein degradation. Twelve more samples had to be excluded from further analysis, because they did not comply with inclusions criteria (6 had a KRAS mutation that was not initially determined as these patients were diagnosed before KRAS sequencing became a prerequisite for anti-EGFR treatment, for 2 tumours cellularity was too low, 2 patients were responders to a first-line combination of anti-EGFR and chemotherapy so that we could not determine the specific response to the anti-EGFR, and 2 tumours were not CRC). Thus, 34 samples were kept for further analysis.
Reverse phase protein arrays
Samples were disrupted in Laemmli buffer (50 mM Tris (pH=6.8), 2% SDS, 5% glycerol, 2 mM DTT, 2.5 mM EDTA, 2.5 mM EGTA, 1 × HALT Phosphatase inhibitor (Thermo Scientific, Illkirch, France 78420), Protease inhibitor cocktail complete MINI EDTA-free (Roche, Meylan cedex, France 1836170, 1 tablet per 10 ml), 2 mM Na3VO4 and 10 mM NaF), using a Precellys (Bertin, Montigny le Bretonneux, France). Extracts were then boiled for 10 min at 100 °C, sonicated to reduce viscosity and centrifuged 10 min at 15 000 r.p.m. The supernatant was collected and stored at −80 °C. Protein concentration was determined (BCA reducing agent compatible kit, ref 23252, Pierce, Thermo Scientific). Samples were printed onto nitrocellulose covered slides (Supernova, Grace Biolabs, Bend, OR, USA) using a dedicated arrayer (2470 arrayer, Aushon Biosystems, Billerica, MA, USA). Five serial dilutions, ranging from 1500 to 94 μg ml−1, and three technical replicates per dilution were printed for each sample. Arrays were labelled with 86 specific antibodies (see Supplementary Table 1 for a complete list of antibody references) or without primary antibody (negative control), using an Autostainer Plus (Dako, Les Ulis, France). Briefly, slides were incubated with avidin, biotin and peroxidase-blocking reagents (Dako) before saturation with TBS containing 0.1% Tween-20 and 5% BSA (TBST–BSA). Slides were then probed overnight at 4 °C with primary antibodies diluted in TBST–BSA. After washes with TBST, arrays were probed with horseradish peroxidase-coupled secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) diluted in TBST-BSA for 1 h at RT. To amplify the signal, slides were incubated with Bio-Rad Amplification Reagent (Bio-Rad, Marnes-la-Coquette, France) for 15 min at RT. The arrays were washed with TBST, probed with Alexa647-Streptavidin (Molecular Probes, Thermo Scientific) diluted in TBST–BSA for 1 h at RT and washed again in TBST. For staining of total protein, arrays were incubated 15 min in 7% acetic acid and 10% methanol, rinsed twice in water, incubated 10 min in Sypro Ruby (Invitrogen, Thermo Scientific) and rinsed again. The processed slides were dried by centrifugation and scanned using a GenePix 4000B microarray scanner (Molecular Devices, Sunnyvale, CA, USA). Spot intensity was determined with MicroVigene software (VigeneTech Inc., Carlisle, MA, USA). All primary antibodies used in RPPA have been previously tested by western blotting to assess their specificity for the protein of interest.
Samples with low overall signal or with aberrant dilution curves, which are often indicative of protein degradation, were discarded during quality control. For each antibody, the median signal intensity was at least three times higher than the background array without primary antibody. Raw data were normalised using Normacurve (Troncale et al, 2012), which normalises spot-wise for a negative control slide (labelled without any primary antibody, provides the fluorescent background) and a slide labelled with a total protein stain (serves as a loading control). Next, Normacurve draws the antibody response curve for each array. Each sample, including the five serial dilutions and the replicates, is aligned onto this curve to calculate one normalised value per sample. These normalised values are then used for statistical analysis.
Statistical analysis
Response to anti-EGFR treatment was determined using RECIST criteria (Therasse et al, 2000). Continuous variables are described as mean and s.d.’s and qualitative data are presented as a number and percentage of sample size. The association between clinical variables and proteins was determined by Wilcoxon or Kruskal–Wallis tests. Association between discrete variables was tested using χ2- or Fisher’s exact test. Hierarchical clustering was performed using Ward metrics and Pearson correlation and represented as a heatmap.
Protein expression was divided in high expression and low expression with the cutoff at the median expression level.
The RECIST criteria were restricted to a dichotomous output, where complete or partial response were considered as a response and stabilisation or progression as a non-response. Univariate logistic regression analysis was performed and odds ratios (ORs) were calculated to measure the association between the expression level of a protein and the chance to respond to treatment. A multivariate logistic regression analysis was performed including significant proteins (P<0.05) with a stepwise procedure.
Overall survival is defined as the time between diagnosis of the metastasis and the date of death. Patients still alive at the moment of analysis were censured at the date of last follow-up. Survival curves were estimated using Kaplan–Meier and compared with log-rank tests.
Univariate and multivariate Cox proportional hazard models were performed to determine the variables that impacts OS. Only variables with a significant P-value (P<0.05) were included in a multivariate stepwise procedure using the Cox model.
P-values below 0.05 were considered significant. All the analyses were performed using R software version 3.3.0 (R Core Team, 2015).
Results
Patient characteristics and response to anti-EGFR therapy according to primary tumour location
Final RPPA data were obtained for 34 CRCs and 86 antibodies. The antibodies were selected according to signalling pathways that have been put forward as being involved in EGFR signalling and anti-EGFR resistance (various RTKs, PI3K/Akt pathway and MAPK pathways) or more generally in CRC carcinogenesis (Wnt/Notch) and chemotherapy response (apoptosis, cell proliferation, DNA repair and angiogenesis). Clinical characteristics are summarised in Table 1 and antibodies are listed in Supplementary Table 1. Median follow-up of these 34 patients was 15.5 months from the beginning of anti-EGFR treatment (range: 3–45.6 months). All patients received anti-EGFR as second- or third-line treatment, except for 4 patients who received anti-EGFR in combination with chemotherapy as a first-line treatment but did not respond. All patients of this cohort were thus shown to be chemoresistant, and any observed response to therapy could be attributed solely to the addition of anti-EGFR treatment. In all, 11 patients showed a partial response, 11 patients showed stabilisation and 12 patients showed disease progression. No complete response was observed. Among the 11 patients with stabilisation, the median time to progression was 8 months (range: 3–13 months) vs 16 months (range: 5–46 months) in patients with partial response and 2 months (range: 1–3 months) in patients with progressive disease. Given the small sample size of patients with stabilisation, we did not perform a further categorisation of stabilised patients according to duration of stabilisation, which would limit the significance of the results. In all, 5 patients showed a PIK3CA hot spot activating mutation (3 patients have p.E545K, 1 patient p.E542K and 1 patient p.N1044K) and 1 other patient a BRAF V600E mutation. Biopsies were obtained before initiation of the anti-EGFR therapy.
We analysed whether the response to treatment, measured according to RECIST criteria, was associated with any of the clinical parameters. The RECIST score was not significantly different according to sex (men vs female), age (<50 vs ⩾50) or the molecule of anti-EGFR treatment (cetuximab vs panitumumab) that was administered. As expected, the absence of objective response was associated to worse overall survival (data not shown). Interestingly, and in accordance with recent findings (Moretto et al, 2016; Tejpar et al, 2016; Holch et al, 2017) the response to anti-EGFR treatment was better in left-sided CRC (descending colon and rectum), where 46% of patients show partial response, than in right-sided CRC (ascending colon), where none of the patients shows partial response (P=0.03).
Protein expression according to patient and tumour characteristics
We studied whether certain proteins, measured by RPPA, were associated with clinical parameters. None of the measured proteins showed a significant association with sex, age or the number of metastatic sites (1 vs ⩾2). Next, we compared the protein profiles of left-sided vs right-sided CRC. Out of 86 protein analysed, 76 proteins are differentially expressed or activated between the two locations (P<0.05), demonstrating an important difference in terms of signalling pathway activation. The phosphorylated proteins that are differentially expressed between left- and right-sided CRC are enriched in HER2/HER3 signalling (P=0.014) and in the Wnt pathway (P=0.018; Figure 1A), which both seem to be more activated in left-sided CRC as previously suggested (Kim et al, 2015; Figure 1B and C).

Differences in pathway activation between left-sided vs right-sided CRC. (A) Phosphorylated proteins that are differentially expressed (P<0.05) between left-sided and right-sided CRC were analysed using Ingenuity Pathway Analysis and found to be enriched in ErbB and Wnt signalling. Enrichment is calculated against the list of analysed proteins. The indicated threshold of −log (P-value)=1.3 corresponds to P=0.05. Expression data were overlaid on a schematic representation of the ErbB (B) and Wnt pathways (C) showing a higher activation of all measured proteins in left-sided colon. The red colour gradient of the proteins represents the fold change between left- and right-sided CRC, with a darker colour indicating a greater fold-change. White proteins are part of the pathway but have not been analysed in this project.
Protein biomarkers predictive for response to anti-EGFR therapy
Using the RECIST criteria, we searched for (phospho-) proteins associated with response to anti-EGFR antibodies that could thus constitute potential predictive biomarkers. Because of small group sizes and to better identify biomarkers the most predictive of response to anti-EGFR, we compared partial response (n=11) vs stabilisation+progression (n=23). Higher levels of Phospho-Akt (Ser473) (P=0.01), HER2 (P=0.03), PKC delta (P=0.03), phospho-HER4 (P=0.04), phospho-p70S6kinase (P=0.05) and 4EBP1 (P=0.05) are associated with response to treatment in univariate analyses (Figure 2). In addition, several proteins show a trend towards significance: higher levels of phospho-EGFR (P=0.06), FGFR4 (P=0.06), phospho-GSK3 (P=0.07) and p53 (P=0.07) are associated with a better response to treatment (Figure 2). Multivariate analyses allow evidencing phospho-Akt (Ser473) as the dominating biomarker for response (OR=5.5, CI 95% (1.6; 34.2)).

Distribution of (phospho-)proteins that are differentially expressed according to the response to anti-EGFR treatment as measured with the RECIST criteria: comparison of stable disease+progressive disease vs partial response (PR). No complete response was observed in our study. P-values are indicated above each comparison. Only protein biomarkers with a P-value of ⩽0.07 are shown. Boxes contain 50% of samples, horizontal line represents the median and isolated dots represent outliers.
Prognostic factors associated with survival
Next, we studied which characteristics were associated with overall survival, defined as the time between the diagnosis of the metastasis and death. Using unsupervised hierarchical clustering, the 86 analysed proteins allow a separation of the 34 CRCs into two clearly distinguishable groups (Supplementary Figure 1). The two clusters do not separate the patients according to response, location of the primary tumour (left- or right-sided, or rectum vs colon) or the centre of origin of the samples. In addition, the two clusters do not display a significant difference in survival (data not shown). Left-sided CRC seem to have a better overall survival compared to right-sided CRC (survival rate at 24 months of 78% (CI 95% (63%; 98%)) and 36% (CI 95% (13%; 99%)) respectively), although the log-rank test is not significant (Supplementary Figure 2).
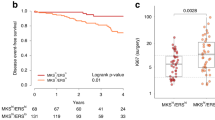
Next, we studied which individual (phospho-)proteins are associated with overall survival. We found that a high expression of EGFR (P=0.01), phospho-EGFR (Tyr1173) (P=0.03) and HER3 (P=0.03) is associated with a better survival (Figure 3). The expression of phospho-EGFR and HER3 proteins is highly correlated (correlation coefficient R=+0.82, P<0.001) and they show identical survival curves (Figure 3B and C). Both proteins also correlate with total EGFR (R=0.62 and 0.61 respectively; P<0.001). Higher expression of FGFR3 (P=0.05), phospho-4EBP1 (P=0.05), p53 (P=0.06) and phospho-HER3 (P=0.06) shows a trend towards an association with better survival, without reaching significance.

(Phospho-)proteins associated with overall survival. Kaplan–Meier curves of overall survival according to expression levels of EGFR (A), phospho-EGFR (Tyr1173) (B) and HER3 (C). Red line: expression higher than the median expression level; blue line: expression lower than the median. The result of the log-rank test is indicated in each graph and the patients at risk over time are indicated below each graph.
Correlation between mutational data, response to treatment and protein expression
In our cohort, a single patient had a BRAF V600E mutation and had progressive disease. Five patients had PIK3CA mutations. From these five patients, three had a partial response; one had a stable disease and one a progressive disease. Although the numbers are too small for statistical analysis, there is thus no indication in our cohort that PIK3CA mutation associates with anti-EGFR resistance. Despite the lack of statistical power, we addressed whether the five PIK3CA-mutated tumours indeed show a higher activation of the Akt pathway than PIK3CA WT tumours. The ratios of phospho-Akt (Thr308)/Akt, of phospho-p70S6K/p70S6K and of P-PKCα/PKCα (a downstream target of PDK1 and mTOR) were slightly increased when PIK3CA is mutated (P=0.11, 0.14 and 0.10, respectively), without reaching significance though (Supplementary Figure 3).
Discussion
Only a fraction of mCRC patients having WT KRAS and NRAS benefit from anti-EGFR treatment, suggesting the presence of additional molecular characteristics leading to primary resistance. Many genetic alterations have been shown to induce resistance in cell lines or xenograft models. Here we hypothesised that all these alterations will ultimately lead to the activation of cell signalling pathways that can be measured at the protein level.
We performed the largest targeted proteomics study published so far in terms of analysed proteins on a small but well annotated cohort of 34 KRAS and NRAS WT mCRC samples from patients that received anti-EGFR therapy, with the aim to identify predictive markers of sensitivity or resistance.
We identified several (phospho-)proteins that are predictive for response to treatment or for overall survival in mCRC patients. Although the observed differences per protein are small and not highly significant due to the small study size, the identified proteins reveal interesting patterns.
Indeed, we show that independently of the line of treatment, patients with higher expression and activation of EGFR and HER3 membrane receptors have a better overall survival. HER3, which lacks a functional kinase domain, heterodimerises with EGFR or with HER2 to produce a potent signalling complex (Jura et al, 2009). Targeting EGFR and HER3 concomitantly is a current lead in CRC (Juric et al, 2015; Temraz et al, 2016). Thus, actively signalling EGFR is associated with a better overall survival in these patients receiving anti-EGFR treatment, probably because these tumours are more dependent on EGFR signalling and thus more sensitive to its inhibition.
If we look more specifically at the response to treatment, as defined by the RECIST criteria, a broader picture appears. The proteins that associate with a better response to therapy are mostly associated with an activation of tyrosine kinases (EGFR, HER2, HER4 and FGFR4) and the downstream signalling through the PI3K/Akt/mTOR pathway (GSK3 and 4EBP1, which are downstream of Akt; p70S6K, which is downstream of mTOR; and PKCΩ, which can be activated by EGFR), thus revealing a complex regulatory network (Figure 4). Thus, patients with activated cell surface receptors (notably EGFR) and PI3K pathway are more likely to respond to anti-EGFR therapy. Our data confirm and extent previously reported observations on the predictive value of phospho-EGFR and phospho-Akt (Van Schaeybroeck et al, 2005; Harle et al, 2015).

Simplified scheme of signalling interaction network between the proteins that are associated with response to anti-EGFR therapy. The red colour gradient reflects the P-value with a darker colour indicating a lower P-value. Only direct and experimentally proven interactions between proteins, described in literature, are shown.
In CRC carrying a mutation in KRAS or NRAS, this mutation confers resistance to anti-EGFR treatment by activating MAPK and PI3K pathways. In our patient cohort of RAS WT CRCs, we observe active EGFR signalling and downstream PI3K pathway activation. Interestingly, we do not identify components of the MAPK pathway as being predictive for response to treatment, suggesting that in RAS WT patients the PI3K pathway is the predominant pathway that explains variability in response to anti-EGFR therapies. Several potential mechanisms could be at the origin of the EGFR activation in these RAS WT tumours. First, the overexpression of EGFR ligands and notably epiregulin and amphiregulin, which activate EGFR, has been associated with a better response to anti-EGFR therapy in RAS WT CRC (Khambata-Ford et al, 2007; Baker et al, 2011; Jonker et al, 2014; Seligmann et al, 2016). Second, mutations in genes such as PIK3CA, PTEN, EGFR and ERBB2, were recently found predictive for anti-EGFR therapy in 31% of RAS WT tumours (Rankin et al, 2016). In our cohort, mutation status was determined for BRAF and PIK3CA. A single patient had a BRAF-mutated tumour and had progressive disease as expected. Indeed, the V600E mutation in the gene that encodes BRAF, which acts downstream of RAS, is known to confer resistance to anti-EGFR therapy and a very poor prognosis of CRC patients (Pietrantonio et al, 2015; Rowland et al, 2015). PIK3CA mutations were previously also suggested to lead to resistance to anti-EGFR therapy (Sartore-Bianchi et al, 2009), although large-scale meta-analyses suggest that this is true only for mutations in exon 20 and not for mutations in exon 9 (De Roock et al, 2010). We detected activating PIK3CA hot spot mutations in five patients: four mutations in exon 9 (three p.E545K and one p.E542K mutation) and one in exon 20 (p.N1044K). From these five patients, three had a partial response (including the patient with the exon 20 mutated tumour), one had a stable disease and one a progressive disease. Although the numbers are too small for statistical analysis, there is thus no indication in our cohort that PIK3CA mutation associates with anti-EGFR resistance. On the contrary, PIK3CA mutations could be one method to activate the PI3K pathway, which we find associated with a better response to therapy.
During the course of our study, large-scale meta-analyses revealed that left-sided and right-sided CRC do not respond similarly to anti-EGFR therapy (Moretto et al, 2016; Tejpar et al, 2016; Holch et al, 2017). We here confirm that left-sided CRC has a better response to anti-EGFR therapy, a tendency towards better survival and a very different profile of protein expression with notably more Wnt and ErbB signalling activation.
In conclusion, we identified activated EGFR and HER3 as biomarkers predictive for a better overall survival in patients treated by anti-EGFR therapy. Response to treatment, on the other hand, was associated with several markers that converge to active EGFR signalling and in particular the PI3K pathway. Validation of these markers by immunohistochemistry on a large panel of samples would therefore be a crucial step forwards to improved patient stratification and personalised medicine in RAS WT CRC.
Change history
05 December 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26 (10): 1626–1634.
American Cancer Society. https://www.cancer.org (accessed March 28, 2017).
Arteaga CL, Engelman JA (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25 (3): 282–303.
Bajpe PK, Prahallad A, Horlings H, Nagtegaal I, Beijersbergen R, Bernards R (2014) A chromatin modifier genetic screen identifies SIRT2 as a modulator of response to targeted therapies through the regulation of MEK kinase activity. Oncogene 27 (10): 588.
Baker JB, Dutta D, Watson D, Maddala T, Munneke BM, Shak S, Rowinsky EK, Xu LA, Harbison CT, Clark EA, Mauro DJ, Khambata-Ford S (2011) Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br J Cancer 104 (3): 488–495.
Bardelli A, Siena S (2010) Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 28 (7): 1254–1261.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Cora D, Di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, Massucco P, Pisacane A, Molinaro L, Valtorta E, Sartore-Bianchi A, Risio M, Capussotti L, Gambacorta M, Siena S, Medico E, Sapino A, Marsoni S, Comoglio PM, Bardelli A, Trusolino L (2011) A molecularly annotated platform of patient-derived xenografts (‘xenopatients’) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1 (6): 508–523.
Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, Sassi F, Cottino F, Migliardi G, Zanella ER, Ribero D, Russolillo N, Mellano A, Muratore A, Paraluppi G, Salizzoni M, Marsoni S, Kragh M, Lantto J, Cassingena A, Li QK, Karchin R, Scharpf R, Sartore-Bianchi A, Siena S, Diaz LA Jr., Trusolino L, Velculescu VE (2015) The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 526 (7572): 263–267.
Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P (2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 27 (5): 663–671.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11 (8): 753–762.
Douillard JY, Rong A, Sidhu R (2013) RAS mutations in colorectal cancer. N Engl J Med 369 (22): 2159–2160.
Harle A, Salleron J, Perkins G, Pilati C, Blons H, Laurent-Puig P, Merlin JL (2015) Expression of pEGFR and pAKT as response-predictive biomarkers for RAS wild-type patients to anti-EGFR monoclonal antibodies in metastatic colorectal cancers. Br J Cancer 113 (4): 680–685.
Heinemann V, Rivera F, O'Neil BH, Stintzing S, Koukakis R, Terwey JH, Douillard JY (2016) A study-level meta-analysis of efficacy data from head-to-head first-line trials of epidermal growth factor receptor inhibitors versus bevacizumab in patients with RAS wild-type metastatic colorectal cancer. Eur J Cancer 67: 11–20.
Holch JW, Ricard I, Stintzing S, Modest DP, Heinemann V (2017) The relevance of primary tumour location in patients with metastatic colorectal cancer: a meta-analysis of first-line clinical trials. Eur J Cancer 70: 87–98.
Jonker DJ, Karapetis CS, Harbison C, O'Callaghan CJ, Tu D, Simes RJ, Malone DP, Langer C, Tebbutt N, Price TJ, Shapiro J, Siu LL, Wong RP, Bjarnason G, Moore MJ, Zalcberg JR, Khambata-Ford S (2014) Epiregulin gene expression as a biomarker of benefit from cetuximab in the treatment of advanced colorectal cancer. Br J Cancer 110 (3): 648–655.
Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J (2009) Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci USA 106 (51): 21608–21613.
Juric D, Dienstmann R, Cervantes A, Hidalgo M, Messersmith W, Blumenschein GR Jr., Tabernero J, Roda D, Calles A, Jimeno A, Wang X, Bohorquez SS, Leddy C, Littman C, Kapp AV, Shames DS, Penuel E, Amler LC, Pirzkall A, Baselga J (2015) Safety and pharmacokinetics/pharmacodynamics of the first-in-class dual action HER3/EGFR antibody MEHD7945A in locally advanced or metastatic epithelial tumors. Clin Cancer Res 21 (11): 2462–2470.
Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359 (17): 1757–1765.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA 3rd, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ (2007) Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 25 (22): 3230–3237.
Kim SE, Paik HY, Yoon H, Lee JE, Kim N, Sung MK (2015) Sex- and gender-specific disparities in colorectal cancer risk. World J Gastroenterol 21 (17): 5167–5175.
Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM 3rd, Perera T, Bertotti A, Trusolino L, Comoglio PM, Boccaccio C (2014) MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res 74 (6): 1857–1869.
Moretto R, Cremolini C, Rossini D, Pietrantonio F, Battaglin F, Mennitto A, Bergamo F, Loupakis F, Marmorino F, Berenato R, Marsico VA, Caporale M, Antoniotti C, Masi G, Salvatore L, Borelli B, Fontanini G, Lonardi S, De Braud F, Falcone A (2016) Location of primary tumor and benefit from anti-epidermal growth factor receptor monoclonal antibodies in patients with RAS and BRAF wild-type metastatic colorectal cancer. Oncologist 21 (8): 988–994.
Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi I, Lonati V, Ghilardi M, de Braud F, Barni S (2015) Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer 51 (5): 587–594.
Price T, Kim TW, Li J, Cascinu S, Ruff P, Suresh AS, Thomas A, Tjulandin S, Guan X, Peeters M (2016) Final results and outcomes by prior bevacizumab exposure, skin toxicity, and hypomagnesaemia from ASPECCT: randomized phase 3 non-inferiority study of panitumumab versus cetuximab in chemorefractory wild-type KRAS exon 2 metastatic colorectal cancer. Eur J Cancer 68: 51–59.
R Core Team (2015) A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria.
Rankin A, Klempner SJ, Erlich R, Sun JX, Grothey A, Fakih M, George TJ Jr., Lee J, Ross JS, Stephens PJ, Miller VA, Ali SM, Schrock AB (2016) Broad detection of alterations predicted to confer lack of benefit from EGFR antibodies or sensitivity to targeted therapy in advanced colorectal cancer. Oncologist 21: 1306–1314.
Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, Sorich MJ (2015) Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer 112 (12): 1888–1894.
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2009) PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 69 (5): 1851–1857.
Seligmann JF, Elliott F, Richman SD, Jacobs B, Hemmings G, Brown S, Barrett JH, Tejpar S, Quirke P, Seymour MT (2016) Combined epiregulin and amphiregulin expression levels as a predictive biomarker for panitumumab therapy benefit or lack of benefit in patients with RAS wild-type advanced colorectal cancer. JAMA Oncol; epub ahead of print 11 February 2016; doi:10.1001/jamaoncol.2015.6065.
Song N, Liu S, Zhang J, Liu J, Xu L, Liu Y, Qu X (2014) Cetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cells. Int J Mol Sci 15 (4): 5838–5851.
Tejpar S, Stintzing S, Ciardiello F, Tabernero J, Van Cutsem E, Beier F, Esser R, Lenz HJ, Heinemann V (2016) Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: retrospective analyses of the CRYSTAL and FIRE-3 trials. JAMA Oncol; epub ahead of print 10 October 2016; doi:10.1001/jamaoncol.2016.3797.
Temraz S, Mukherji D, Shamseddine A (2016) Dual targeting of HER3 and EGFR in colorectal tumors might overcome anti-EGFR resistance. Crit Rev Oncol Hematol 101: 151–157.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216.
Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, De Palma R, D'Aiuto E, Berrino L, Bianco R, Ciardiello F (2013) Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res 19 (24): 6751–6765.
Troncale S, Barbet A, Coulibaly L, Henry E, He B, Barillot E, Dubois T, Hupe P, de Koning L (2012) NormaCurve: a SuperCurve-based method that simultaneously quantifies and normalizes reverse phase protein array data. PLoS One 7 (6): e38686.
Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360 (14): 1408–1417.
Van Emburgh BO, Sartore-Bianchi A, Di Nicolantonio F, Siena S, Bardelli A (2014) Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol Oncol 14 (14): 00096–00099.
Van Schaeybroeck S, Karaiskou-McCaul A, Kelly D, Longley D, Galligan L, Van Cutsem E, Johnston P (2005) Epidermal growth factor receptor activity determines response of colorectal cancer cells to gefitinib alone and in combination with chemotherapy. Clin Cancer Res 11 (20): 7480–7489.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K, Fukuoka M, Varella-Garcia M, Nakagawa K, Janne PA (2011) Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 3 (99): 99ra86.
Acknowledgements
We thank Stéphane Liva and Patrick Poullet for bioinformatics support. This work was financed by the Institut Curie Evaluation Committee of Translational Research (CEST) and Institut Carnot Curie-Cancer 2013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lièvre, A., Ouine, B., Canet, J. et al. Protein biomarkers predictive for response to anti-EGFR treatment in RAS wild-type metastatic colorectal carcinoma. Br J Cancer 117, 1819–1827 (2017). https://doi.org/10.1038/bjc.2017.353
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.353

{kind=link}
{kind=link}
{kind=link}