
Abstract
Background:
A substantial fraction of familial colorectal cancer (CRC) and polyposis heritability remains unexplained. This study aimed to identify predisposing loci in patients with these disorders.
Methods:
Homozygosity mapping was performed using 222 563 SNPs in 302 index patients with various colorectal neoplasms and 3367 controls. Linkage analysis, exome and whole-genome sequencing were performed in a family affected by microsatellite stable CRCs. Candidate variants were genotyped in 10 554 cases and 21 480 controls. Gene expression was assessed at the mRNA and protein level.
Results:
Homozygosity mapping revealed a disease-associated region at 1q32.3 which was part of the linkage region 1q32.2–42.2 identified in the CRC family. This includes a region previously associated with risk of CRC. Sequencing identified the p.Asp1432Glu variant in the MIA3 gene (known as TANGO1 or TANGO) and 472 additional rare, shared variants within the linkage region. In both cases and controls the population frequency was 0.02% for this MIA3 variant. The MIA3 mutant allele showed predominant mRNA expression in normal, cancer and precancerous tissues. Furthermore, immunohistochemistry revealed increased expression of MIA3 in adenomatous tissues.
Conclusions:
Taken together, our two independent strategies associate genetic variations in chromosome 1q loci and predisposition to familial CRC and polyps, which warrants further investigation.
Similar content being viewed by others


Main
Family history is a major risk factor in colorectal cancer (CRC) aetiology. Familial aggregation of disease is observed in up to 25% of the cases and estimates from twin studies indicate that inherited genetic factors contribute to the development of CRCs in approximately 15% of cases (Kerber et al, 2005; Mucci et al, 2016). It is generally accepted that a substantial proportion of CRC incidence is due to predisposition to non-malignant CRC lesions (e.g., adenomas, serrated lesions). Indeed, first-degree relatives of patients with colorectal adenomas have an increased risk of developing colorectal adenocarcinomas (Johns and Houlston, 2001; Tuohy et al, 2014).
Only up to 14% of CRC predisposition can be attributed to the known Mendelian-inherited syndromes and common gene variants, suggesting that a substantial fraction of this heritability remains unexplained (Aaltonen et al, 2007; Dunlop et al, 2013; Jiao et al, 2014). In support of this, linkage studies in CRC families have identified disease-associated chromosomal areas where predisposing genes have not yet been identified (Wiesner et al, 2003; Kemp et al, 2006; Neklason et al, 2008; Middeldorp et al, 2010). Similarly, some regions, such as chromosome 1q41 (rs6691170, rs6687758 and rs11118883), identified in genome-wide association studies (GWAS) still have not led to the identification of functional CRC variants (Houlston et al, 2010; Spain et al, 2012; Zhang et al, 2014). Furthermore, one study measuring runs of homozygosity in search for recessive disease-causing loci, did not identify novel CRC predisposing loci (Spain et al, 2009). In recent years, advances in sequencing technologies have made it possible to identify germline susceptibility loci that were previously not encountered in linkage and association studies. For example, using a combination of sequencing and linkage studies germline mutations were identified in two DNA-polymerase genes (POLE and POLD1) (Palles et al, 2013).
In this study, we searched for germline variants involved in colorectal neoplasm susceptibility using two independent genomic strategies. Homozygosity mapping analysis was conducted in a cohort of 302 index patients with various colorectal neoplasms, ranging from few to hundreds of polyps and 3367 controls. In parallel, a combination of linkage analysis and sequencing was performed in individuals from a large family with an extensive history of microsatellite stable CRC. Both strategies identified an association of chromosome 1q loci with these diseases.
Materials and methods
Ethics
The study was approved by the Medical Ethical Committee of the Leiden University Medical Center, Leiden, The Netherlands (protocol P01–019). Informed consent was given for further research at initial blood withdrawal for the homozygosity mapping cohort, family 68 and the MIA3 genotyping cohorts. Anonymised tissue samples were handled according to the medical ethical guidelines described in the Code Proper Secondary Use of Human Tissue established by the Dutch Federation of Medical Sciences (https://www.federa.org/) and informed consent was waived for the use of these samples by the Medical Ethical Committee (protocol P01–019).
CytoSNP array genotyping
A cohort of patients with multiple adenomas, previously described by (Hes et al, 2014), and patients with <10 adenomas or a phenotype with predominantly serrated lesions, were included in this study. The clinical characteristics of the cohort are presented in Supplementary Table 1. All cases were negative for known pathogenic variants in the MMR genes, APC (including mosaic APC), POLE, POLD1, MUTYH or NTHL1. A total of 343 cases were genotyped using the Illumina HumanCytoSNP-12 v2.0 or v2.1 BeadChips (Illumina Inc., San Diego, CA, USA). For controls we used genotypes of 3367 individuals from the LifeLines cohort study (Stolk et al, 2008; Dolmans et al, 2011). A total of 222 563 SNPs in 302 cases and 3367 controls passed quality control. The quality control procedure is described in Supplementary Methods.
Homozygosity mapping
SNP selection and homozygosity mapping analysis is described in Supplementary Methods. In total 78 582 SNPs were included in the analysis. Runs of homozygosity were compared between cases and controls using the Fisher‘s exact test (P<1.2 × 10−6).
Genome-wide association study
A quantile–quantile (Q–Q) plot of the observed and expected P-values indicated differential genotyping between cases and controls. A genomic inflation factor (based on median χ2) λ=1.23378 was applied to correct for the population bias between cases and controls. Genotype prevalence of 222 563 SNPs was compared between cases and controls using a χ2 allelic test with 1 degree of freedom. Odds ratio and its 95% confidence interval was also computed. Multiple test correction was done using false discovery rate estimation (Benjamini and Yekutieli, 2001).
Family 68 characterisation
Clinicopathological data of family 68 family members were obtained from the national registry of The Netherlands Foundation for the Detection of Hereditary Tumours (http://www.stoet.nl) and are shown in Supplementary Table 2. Peripheral blood was collected from 16 members of family 68. DNA was extracted using standard techniques. Index patient 68-01 had been screened and was found to be negative for known pathogenic variants in the MMR genes, APC, POLE, POLD1, MUTYH and NTHL1.
Linkage analysis
Fifteen family members of family 68, including one unaffected spouse, were genotyped using the GeneChip Mapping 10 K Xba 142 array (Affymetrix Inc., Santa Clara, CA, USA). Data was previously described (Middeldorp et al, 2010). SNPs with a call rate below 90% were excluded from further analysis. A total of 9131 SNPs had a valid annotation. Genotypes incompatible with the family relations were removed using pedcheck. Non-parametric linkage analysis was performed with MERLIN (Abecasis et al, 2002) using S-all scoring (Whittemore and Halpern, 1994) with the exponential model (Kong and Cox, 1997). Patients diagnosed with CRC before 60 years or with colorectal polyps before 55 years were considered affected. Other family members were classified as unaffected.
Exome and whole-genome sequencing
Exome sequencing was performed on family 68 DNA samples (68-01, 68-08, 68-11, 68-15 and 68-17) using HiSeq (Illumina) or Complete Genomics (Complete Genomics Inc., Mountain View, CA, USA) sequencing technology (Carnevali et al, 2012) as described in Supplementary Methods. Samples (68-07, 68-11 and 68-15) were sent to Macrogen (Macrogen Europe, Amsterdam, The Netherlands) for whole-genome sequencing (Supplementary Methods). Variants with a population frequency>0.01 (in 1000Genomes, ExAC, ESP or GoNL databases) were excluded. For the analysis of the exome sequencing data, we selected variants predicted to be coding or affecting splice-sites. In the whole-genome analysis, we prioritised the non-coding variants which were present within the linkage peak based on regions under strong negative selection, as previously described (Khurana et al, 2013).
MIA3 p.Arg1432Glu genotyping
The MIA3 c.4296T>A, p.Asp1432Glu variant was validated by standard bidirectional Sanger sequencing. A KASPar assay was designed for the MIA3 p.Asp1432Glu variant by KBioscience (LGC Genomics, Hoddesdon, UK). Leucocyte or normal colon tissue derived DNA was screened of 1477 CRC patients, 445 polyposis patients and 1604 population-based controls (anonymous blood donors) from The Netherlands and 1098 CRC patients and 1459 healthy controls from the Czech Republic (Pardini et al, 2013). All carriers were validated by Sanger sequencing and the existence of a common ancestor was excluded using STR profiling (Supplementary Methods). Additionally, MIA3 genotypes were extracted from a European cohort of 7534 CRC patients and 18 417 healthy controls that were genotyped using OmniExpressExome BeadChip 8v1.1 or 8v1.20 (Illumina) (Timofeeva et al, 2015).
Somatic MIA3 mutation analysis
Tumour DNA of 47 microsatellite stable CRCs was screened for somatic MIA3 mutations (Supplementary Table 3). Paired tumour-normal DNA samples of MIA3 p.Asp1432Glu carriers (68-1 and both carriers identified with screening) were screened for second hits in the MIA3 gene. Sequencing was performed with a custom multiplex PCR on the Ion Torrent Personal Genome Machine sequencer (Life Technologies, Grand Island, NY, USA) as described in Supplementary Methods. Synonymous and intronic variants were excluded as well as variants present in population frequency databases (in 1000Genomes, ExAC, ESP or GoNL).
For comparison, the somatic mutation data from 915 colorectal adenocarcinoma exomes (Muzny et al, 2012; Seshagiri et al, 2012; Giannakis et al, 2016) was retrieved from The cancer genome atlas (TCGA) using cBioPortal (Cerami et al, 2012; Gao et al, 2013).
Quantitative real-time reverse transcriptase PCR
mRNA was isolated from fresh frozen tissue samples of 18 normal tissues, 32 colorectal adenomas and 18 colorectal adenocarcinomas (Supplementary Table 4). mRNA isolation and quantitative real-time reverse transcriptase PCR were performed as previously described (Lips et al, 2008; van Roon et al, 2013). MIA3 mRNA expression of the full-length isoform (primer sequences available upon request) was normalised to housekeeping genes CPSF6 and HNRNPM (van Roon et al, 2013). Group differences were tested with one-way ANOVA.
Allele-specific expression
Formalin-fixed, paraffin-embedded tissue blocks of MIA3 p.Asp1432Glu carriers were collected (caecum carcinoma and normal tissue; two serrated lesions, two adenomas and normal colon tissue). Total nucleic acid was extracted using the Tissue Preparation System (Siemens Healthcare Diagnostics, Tarrytown, NY, USA) (van Eijk et al, 2013). Nucleic acid was treated with DNaseI (Fermentas, Thermo Fisher Scientific, Waltham, MA, USA) and converted into cDNA using the High-Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Allele-specific expression analysis of the MIA3 variant was performed using 2 μl 25x diluted cDNA as template for the KASPar assay. Paired DNA was analysed to determine baseline amplification differences between the two alleles. Using the Cq values obtained for both alleles, the allelic dosage was calculated similarly to the Pfaffl method for relative gene expression (Pfaffl, 2001).
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissues blocks were available of 82 patients from the FACTS study (Hennink et al, 2015) (which included 18 serrated lesions and 97 adenomas with low-grade dysplasia) and a series of 15 anonymous colorectal adenocarcinomas. Formalin-fixed, paraffin-embedded and fresh frozen tissue sections of MIA3 p.Asp1432Glu carriers were stained. MIA3 immunohistochemistry was performed as described in Supplementary Methods. The intensity of MIA3 cytoplasmic staining was classified as negative-weak or moderate-strong. Normal mucosa was analysed when present in the tissue sections. MIA3 gradient expression was scored: maximal staining at the base of the crypts and decreasing towards the lumen (top-down), maximal staining at the lumen and decreasing toward the base of the crypts (down-top) or homogeneous staining across lumen and crypt (no gradient). Differences between tissue types were compared using a Fisher’s exact test.
Results
Genotyping and homozygosity mapping in colorectal neoplasm cases
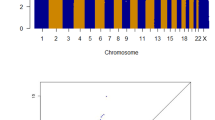
We genotyped germline DNA samples from index patients affected with colorectal neoplasms ranging from few to hundreds of polyps (polyposis), with and without CRC. After sample and SNP quality control procedures and removal of ethnic outliers, the genotypes for 222 563 SNPs from 302 cases and 3367 controls were included. We removed SNPs with a MAF<0.05 or SNPs in linkage disequilibrium, resulting in 80 907 remaining SNPs. Using PLINK, we searched for runs of homozygosity containing ⩾15 homozygous SNPs and spanning a region of at least 1.5 Mb. A total of 40 849 runs of homozygosity were identified in the cases and controls, with an average size of 2.73 Mb. Per individual a similar number of runs of homozygosity (10.4 vs 11.2) and a cumulative length of homozygosity (average 28.7 vs 30.5 Mb), were observed in cases and controls, respectively. On chromosome region 1q32.3, runs of homozygosity were overrepresented in cases compared to controls (2.0 vs 0.1%; P=0.0001), although not reaching genome-wide significance after correction for multiple testing (Figure 1). This 1 Mb region of homozygosity (chr1:211 265 284–212 357 017), bounded by SNPs rs1338348 and rs11119874, was identified in six patients diagnosed with multiple polyps before 56 years of age; five out of six had >10 neoplastic lesions (Supplementary Table 5). This region contains 10 coding genes and 2 small non-coding RNAs (UCSC gene database). No individual SNPs, including chromosome 1q SNPs rs11118883 and rs6687758, reached global significance in a GWAS analysis utilising the same SNPs.

Overrepresentation of runs of homozygosity located on chromosome 1q32.3 in patients with colorectal neoplasms. (A) Frequency of the runs of homozygosity on chromosome 1; cases (blue) and controls (pink). Overrepresented homozygosity in cases is indicated with a red box. (B) Overlapping runs of homozygosity of six cases (blue bars) at chromosomal locus 1q32.3.
Clinical characterisation and linkage analysis in family 68
In parallel to the homozygosity mapping analysis we performed linkage analysis in a family not included in the homozygosity mapping cohort. Updated clinical follow-up information warranted the reanalysis of a previously described Amsterdam criteria I fulfilling family affected with microsatellite stable CRCs without a known causative mutation (Middeldorp et al, 2010). Fifteen individuals of this family (68-01 until 68-15) were genotyped using Affymetrix 10 K SNP arrays (Figure 2). For non-parametric linkage analysis only individuals presenting with early-onset colorectal neoplasms (⩾1 adenoma before 55 years and/or CRC before 60 years) were considered affected. All affected family members developed less than 10 polyps. The clinical presentation of the late-onset patients included the diagnosis of CRC in an elderly patient at 88 years of age (68-13) and the identification of 1–2 adenomas during periodic colonoscopy screenings in three relatives, aged 56–60 years (68-02, 68-06 and 68-09). Furthermore, the linkage analysis included one family member without a history of polyps or cancer at 59 years of age and one unaffected spouse. We identified a region of suggested linkage (logarithm of odds (LOD)>2.5; maximum LOD score=2.75) on chromosome 1q32.2-q42.2 (Figure 3). No additional linkage regions were identified (LOD>0.8). The linkage region spans 21.1 Mb (chr1:210 940 342–232 031 815) and overlaps with the region of homozygosity described above, located on chromosome 1q32.3. The whole region is predicted to contain 137 coding genes and 48 small non-coding RNAs (UCSC gene database).

Pedigree of family 68. Filled symbol, colorectal cancer. Filled quarter, ⩾1 adenoma. Black, early-onset colorectal neoplasms (adenoma<55 years; CRC<60 years). Grey, late-onset colorectal neoplasms. Numbers indicate individuals whose DNA was available.

Non-parametric linkage analysis in family 68. Logarithm of odds scores of chromosome 1 (in centiMorgan) of the non-parametric linkage analysis of 15 individuals using the exponential model.
Germline sequencing in family 68
To further investigate the chr1q loci identified with homozygosity mapping and linkage analysis, we focussed on the largest region, the linkage region in family 68. In search of disease-associated variants, exome sequencing was performed on DNA from five affected family members (68-1, 68-8, 68-11, 68-15 and 68-17). Exonic or predicted splice-site variants with a maximum population frequency below 0.01 were selected. Three variants were shared amongst these five individuals (Table 1), of which only one was located within the linkage peak; the c.4296T>A, p.Asp1432Glu variant in Melanoma Inhibitory Activity protein 3 gene (MIA3, also known as TANGO1 or TANGO). Additional variants identified within the linkage peak were not shared between the individuals (Supplementary Table 6). Segregation analysis, using Sanger sequencing, showed that all family members presenting with early-onset colorectal neoplasms were carriers of the MIA3 p.Asp1432Glu variant, whereas one unaffected relative (68-04), one patient with a single polyp at 60 years of age (68-9) and one unaffected spouse (68-14) were non-carriers. The MIA3 variant was present in three late-onset patients (68-02, 68-06, 68-13).
To investigate the presence of variants in non-coding regions on chromosome 1q32.2-q42.2, whole-genome sequencing was performed in three family members (68-7, 68-11 and 68-15). Of the known CRC risk alleles located on chromosome 1q (rs6691170, rs6687758 or rs11118883) all individuals carried the non-risk alleles with one exception, individual 68-11 was heterozygous for the rs6691170 allele. Next, heterozygous variants shared amongst all three individuals and with a maximum population frequency of 0.01, were extracted from within the linkage peak region. A total of 473 variants were identified, 51% were single nucleotide variants and 49% insertion or deletions (81% in genetic repeat sequences). In total 33 variants were located within the homozygosity mapping region located on chromosome 1q32.3. Within 1 Mb of the MIA3 gene 45 variants were identified (chr1:221 791 444–223 841 354). In addition, one exonic variant was found (OBSCN: NM_001098623 c.4523T>A, p.Val1508Asp), which was not covered in the exome sequencing. Subsequently, we investigated variants located in regions under strong negative selection, i.e. non-coding regions harbouring an increased fraction of rare variants comparable to the coding region, which are subdivided into ultrasensitive (e.g., BRF2-binding site) and sensitive regions (binding sites of some chromatin and general transcription factors). One variant, rs529680452, was identified in a sensitive non-coding element; in the binding domain of histone acetyltransferase KAT2A located in the 5′ untranslated region of the DTL gene (Supplementary Table 7). Thus, the MIA3 p.Asp1432Glu variant seemed the most promising candidate for the colorectal neoplasm predisposition in this family and was selected for further investigation.
Prevalence of the MIA3 p.Asp1432Glu variant
The MIA3 p.Asp1432Glu variant is present at low frequencies in public genotype databases; 0.01% the in ESP database (1/5997), 0.05% in the ExAC database (55/59 662) and 0.1% in the Genome of The Netherlands project (1/498). We performed or collected additional genotyping data of 10 554 cases and 21 480 controls. Analysis over all cohorts revealed an allele frequency of 0.02% in both cases and controls; separate frequencies per cohort are shown in Supplementary Figure 1. KASPar genotyping of the Dutch CRC cohort identified two additional carriers of the p.Asp1432Glu variant. No carriers were found in the cases used for homozygosity mapping.
Clinical characterisation MIA3 p.Asp1432Glu carriers
STR profiling provided no evidence of a recent common ancestor between the 68-01 and the other two carriers. One patient has a positive family history of both CRC and adenomas and has a confirmed germline deletion in MSH2. This patient developed a microsatellite instability-high (MSI-H) caecum tumour at age 44. The other patient had no germline mutations in CRC genes and no known family history of disease. The patient developed microsatellite stable rectal cancer at 40 years and underwent a hemicolectomy of the left-sided colon and rectum at age 56 after developing >20 polyps (both adenomas and serrated lesions).
MIA3 somatic mutations
We performed targeted somatic mutation screening of MIA3 in tumour DNA from p.Asp1432Glu MIA3 carriers. In the MSI-H tumour a somatic mutation was present (c.55701G>A, p.Gly1857Asp). No second hits in MIA3 were detected in the other tumour samples. In addition, MIA3 was screened in 47 familial microsatellite stable CRCs. Somatic non-synonymous MIA3 mutations were observed in 6.4% of the tumours (Supplementary Table 8). Our findings are in line with the somatic mutation frequency of 4.5% in 915 colorectal adenocarcinomas from TCGA.
MIA3 mRNA and allele-specific expression
Using quantitative real-time PCR, MIA3 expression was quantified in mRNA isolated from normal tissues, colorectal adenomas and colorectal adenocarcinomas. High variability in expression was observed within each group and showed no significant difference in expression between the groups (P=0.684; Figure 4A). We investigated the allele-specific expression of the MIA3 variant in tissues derived from p.Asp1432Glu MIA3 carriers. mRNA expression of the mutant allele was three times higher in normal tissue samples and more than four times higher in colorectal neoplasms (Figure 4B).

mRNA and protein expression of MIA3 in colon tissues. (A) mRNA expression of MIA3 (relative to housekeeping genes CPSF6 and HNRNPM) in colonic tissues. (B) The p.Asp1432Glu variant was higher expression than the wild-type allele in both normal colonic tissue and colon lesions. (C) A significant increase in protein expression is observed in adenomas and a significant decrease in carcinomas, compared to normal colorectal tissue sections. (D) Gradient expression of MIA3 protein was more frequent in serrated lesions and adenomas compared to carcinomas. Serrated lesions show higher expression basally, while adenomas had higher expression on the luminal side of the polyp. *P⩽0.05, **P⩽0.01.
MIA3 protein expression
To better understand the potential role of MIA3, we examined MIA3 expression in precancerous colorectal lesions and colorectal carcinoma tissue sections. A general decrease in MIA3 expression was observed in adenocarcinomas compared to normal mucosa (13 vs 46% moderate-strong staining; P=0.04; Figure 4C). Notably, higher MIA3 expression was seen in colorectal polyps, 67% of serrated lesions and 70% of adenomas showed moderate-strong staining (P=0.18, P=0.006, respectively, compared to normal mucosa). A gradient in MIA3 expression was observed in polyps (representative tissue sections in Supplementary Figure 2), while no gradient was present in normal mucosa. In serrated lesions MIA3 expression was highest at the base of the crypts and decreased towards the lumen (down-top gradient), while in adenomas the inverse gradient was observed (top-down gradient) (Figure 4D).
In tissue sections from p.Asp1432Glu carriers, the MIA3 expression was highly variable but not distinctive from non-carriers. These results mainly show high inter- and intra-patient heterogeneity, independent of the MIA3 p.Asp1432Glu germline variant.
Discussion
To date, genetic association studies have reported many SNPs associated with either CRC or multiple adenoma susceptibility, including rs6691170, rs6687758, rs11118883 located on chromosome region 1q41 (Spain et al, 2012; Theodoratou et al, 2012; Montazeri et al, 2016). In contrast, homozygosity mapping studies have been less successful in the identification of predisposition loci in CRC and other types of cancers, indicating that recessive syndromes might be less frequent in genetic predisposition to cancer (Spain et al, 2009; Enciso-Mora et al, 2010; Hosking et al, 2010; Sud et al, 2015). In this study we found a recurrent region of homozygosity located on chromosome 1q32.3 (chr1:211 265 284–212 357 017) in patients with a mixed early-onset colorectal neoplasm phenotype. For this study we included patients with a large spectrum of cancerous and precancerous colorectal lesions, as known hereditary CRC syndromes also have a highly variable clinical presentation. None of the runs of homozygosity or single SNPs in our GWAS analysis reached significance with the genome-wide thresholds, which mainly reflects a lack of power and underscores the need for larger sample sizes. In a highly overlapping cohort, two variants significantly associated with disease, rs3802842 (chr11q23) and rs4779584 (chr15q13), however, only 16 SNPs were tested (Hes et al, 2014). Hence, despite adjustment for multiple testing, the association between early-onset colorectal neoplasm risk and chromosome 1q32.3 in susceptibility to multiple colorectal neoplasms warrants further investigation.
Furthermore, a linkage peak was identified in 15 individuals from a large family with an extensive history of microsatellite stable CRC and polyps located on chromosome 1q32.2–42.2. As an individual finding the identification of the linkage region in a single family or the region of homozygosity in the colorectal neoplasm cohort might not provide compelling evidence for the involvement of this region. However, together the linkage region, the 21.1 Mb region of homozygosity and previously described predisposing and protective CRC risk alleles (Spain et al, 2012) located in a highly overlapping region are suggestive of a broad-based association of chr1q and CRC predisposition. Therefore we decided to further investigate this association by focussing on the broadest, most inclusive region, which was the linkage region identified in family 68.
Although the family presented with a dominant pattern of inheritance, we proceeded with the screening of rare genetics variants in family 68 looking into heterozygous, homozygous and compound heterozygous variants within the linkage region. Based on exome sequencing results, the most likely candidate variant for predisposition was the heterozygous MIA3 c.4296T>A; p.Asp1432Glu variant. MIA3 (also known as TANGO or TANGO1) is a cargo receptor localised to the endoplasmic reticulum and facilitates the trafficking of collagens to the Golgi apparatus (Wilson et al, 2011; Ishikawa et al, 2016). The p.Asp1432 amino acid is located in the cytoplasm in the first out of two coiled-coiled domains of MIA3 (amino acids 1211–1440) (Saito et al, 2011). This region may interact with still unidentified intermediates of this transport carrier complex and we hypothesised that missense variants may stabilise or destabilise these protein interactions. MIA3 was shown to promote angiogenesis and lymphangiogenesis in oral squamous cell carcinoma (Sasahira et al, 2014). Furthermore, patients with MIA3-positive squamous cell carcinomas have a significantly shorter disease-free survival than patients without MIA3 expression (Sasahira et al, 2016). However, downregulation was observed in melanomas, hepatocellular and colon carcinomas (Arndt and Bosserhoff, 2006; 2007; Sasahira et al, 2014). To our knowledge we are the first to describe an increased protein expression in colorectal adenomas, which might indicate a tumourigenic role of MIA3 in the development of these precancerous lesions. In addition, our data showed a tissue gradient in adenomas and the inverse gradient in serrated lesions, possibly correlated to the proliferating cells in these polyps. At the mRNA level no differences were observed between adenomas, carcinomas and normal colon tissue.
No differential protein expression was observed in tissue from carriers and non-carriers of the MIA3 p.Asp1432Glu variant, although only a limited number of tissue blocks were available from carriers. In addition, genotyping data did not support the involvement of this MIA3 variant in CRC predisposition, based on similar allele frequencies in cases and controls. The observed allelic imbalance of the p.Asp1432Glu MIA3 allele in carriers could possibly be explained by the presence of regulatory variants in linkage, leading to the up-regulation of the mutant allele compared to the wild-type allele in these individuals.
To investigate the possibility of non-coding variants underlying CRC predisposition in family 68, whole-genome sequencing was performed. We focussed on heterozygous variants within the linkage region due to the predicted dominant pattern of inheritance in this family. In total 473 heterozygous, shared, rare variants were identified. One additional coding variant, OBSCN p.Arg4213His, was identified in a region not covered by the exome sequencing analysis. The OBSCN gene codes for obscurins, giant cytoskeletal Rho-guanine nucleotide exchange factor proteins, that interact with cytoskeletal calmodulin and titin (Young et al, 2001). The OBSCN gene is frequently mutated in CRC and other cancers, however, to date, research lacks to support an association of germline variants and increased cancer risks (Sjoblom et al, 2006; Balakrishnan et al, 2007; Huhn et al, 2014).
As no regulatory elements of MIA3 have been described, we selected variant located between 1 Mb up- and down-stream of the MIA3 gene and identified 45 variants (Sotelo et al, 2010). None of these variant were located in regions under strong negative selection (Khurana et al, 2013), such as regions important for transcription factor binding, and therefore additional research is needed to investigate the effect of these 45 variants on MIA3 transcription. Of the 473 variants only one was present in a region under strong negative selection, a rare variant (rs529680452) in the binding domain of histone acetyltransferase KAT2A located in the 5′ untranslated region of the DTL gene. Denticleless E3 ubiquitin protein ligase homologue, encoded by the DTL gene, is upregulated in various cancers, including CRC, and it is reported to play a crucial role in carcinogenesis (Cheung et al, 2001; Pan et al, 2006; Ueki et al, 2008; Baraniskin et al, 2012; Kobayashi et al, 2015). Variants in transcription factor binding sites have previously been associated with an increased risk of cancer, the most well studied are long-range enhancers of MYC (Ahmadiyeh et al, 2010; Sotelo et al, 2010). The DTL variant is one out of 33 non-coding variants located in the 1 Mb region identified with homozygosity mapping analysis on chromosome 1q32.3, which are also interesting candidates and warrant for further investigation.
Taken together, this study identifies, with two independent strategies, a genomic region on chromosome 1q which is associated with the predisposition to CRC and multiple polyps. Several other studies have pinpointed this region in CRC genetic association studies. Novel comprehensive approaches are required for the identification of functional variants predisposing to CRC, possibly affecting regulatory elements or non-coding RNAs.
Change history
10 October 2017
This manuscript was initially published online in the wrong issue of British Journal of Cancer. This has now been rectified and a corrigendum has been published.
12 October 2017
An Erratum to this paper has been published: https://doi.org/10.1038/bjc.2017.380
References
Aaltonen L, Johns L, Jarvinen H, Mecklin JP, Houlston R (2007) Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin Cancer Res 13 (1): 356–361.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30 (1): 97–101.
Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, Caswell JL, Beckwith CA, Hills A, Macconaill L, Coetzee GA, Regan MM, Freedman ML (2010) 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci USA 107 (21): 9742–9746.
Arndt S, Bosserhoff AK (2006) TANGO is a tumor suppressor of malignant melanoma. Int J Cancer 119 (12): 2812–2820.
Arndt S, Bosserhoff AK (2007) Reduced expression of TANGO in colon and hepatocellular carcinomas. Oncol Rep 18 (4): 885–891.
Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A (2007) Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res 67 (8): 3545–3550.
Baraniskin A, Birkenkamp-Demtroder K, Maghnouj A, Zollner H, Munding J, Klein-Scory S, Reinacher-Schick A, Schwarte-Waldhoff I, Schmiegel W, Hahn SA (2012) MiR-30a-5p suppresses tumor growth in colon carcinoma by targeting DTL. Carcinogenesis 33 (4): 732–739.
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29 (4): 1165–1188.
Carnevali P, Baccash J, Halpern AL, Nazarenko I, Nilsen GB, Pant KP, Ebert JC, Brownley A, Morenzoni M, Karpinchyk V, Martin B, Ballinger DG, Drmanac R (2012) Computational techniques for human genome resequencing using mated gapped reads. J Comput Biol 19 (3): 279–292.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disc 2 (5): 401–404.
Cheung WM, Chu AH, Chu PW, Ip NY (2001) Cloning and expression of a novel nuclear matrix-associated protein that is regulated during the retinoic acid-induced neuronal differentiation. J Biol Chem 276 (20): 17083–17091.
Dolmans GH, Werker PM, Hennies HC, Furniss D, Festen EA, Franke L, Becker K, van der Vlies P, Wolffenbuttel BH, Tinschert S, Toliat MR, Nothnagel M, Franke A, Klopp N, Wichmann HE, Nurnberg P, Giele H, Ophoff RA, Wijmenga C (2011) Wnt signaling and Dupuytren's disease. N Engl J Med 365 (4): 307–317.
Dunlop MG, Tenesa A, Farrington SM, Ballereau S, Brewster DH, Koessler T, Pharoah P, Schafmayer C, Hampe J, Volzke H, Chang-Claude J, Hoffmeister M, Brenner H, von Holst S, Picelli S, Lindblom A, Jenkins MA, Hopper JL, Casey G, Duggan D, Newcomb PA, Abuli A, Bessa X, Ruiz-Ponte C, Castellvi-Bel S, Niittymaki I, Tuupanen S, Karhu A, Aaltonen L, Zanke B, Hudson T, Gallinger S, Barclay E, Martin L, Gorman M, Carvajal-Carmona L, Walther A, Kerr D, Lubbe S, Broderick P, Chandler I, Pittman A, Penegar S, Campbell H, Tomlinson I, Houlston RS (2013) Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42,103 individuals. Gut 62 (6): 871–881.
Enciso-Mora V, Hosking FJ, Houlston RS (2010) Risk of breast and prostate cancer is not associated with increased homozygosity in outbred populations. Eur J Hum Genet 18 (8): 909–914.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6 (269): pl1.
Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, Sukawa Y, Stewart C, Rosenberg M, Mima K, Inamura K, Nosho K, Nowak JA, Lawrence MS, Giovannucci EL, Chan AT, Ng K, Meyerhardt JA, Van Allen EM, Getz G, Gabriel SB, Lander ES, Wu CJ, Fuchs CS, Ogino S, Garraway LA (2016) Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep 17 (4): 1206.
Hennink SD, van der Meulen-de Jong AE, Wolterbeek R, Crobach AS, Becx MC, Crobach WF, van Haastert M, Ten Hove WR, Kleibeuker JH, Meijssen MA, Nagengast FM, Rijk MC, Salemans JM, Stronkhorst A, Tuynman HA, Vecht J, Verhulst ML, de Vos Tot Nederveen Cappel WH, Walinga H, Weinhardt OK, Westerveld D, Witte AM, Wolters HJ, Cats A, Veenendaal RA, Morreau H, Vasen HF (2015) Randomized comparison of surveillance intervals in familial colorectal cancer. J Clin Oncol 33 (35): 4188–4193.
Hes FJ, Ruano D, Nieuwenhuis M, Tops CM, Schrumpf M, Nielsen M, Huijts PE, Wijnen JT, Wagner A, Gomez Garcia EB, Sijmons RH, Menko FH, Letteboer TG, Hoogerbrugge N, Harryvan J, Kampman E, Morreau H, Vasen HF, van Wezel T (2014) Colorectal cancer risk variants on 11q23 and 15q13 are associated with unexplained adenomatous polyposis. J Med Genet 51 (1): 55–60.
Hosking FJ, Papaemmanuil E, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JA, Allan JM, Taylor M, Tomlinson IP, Greaves M, Houlston RS (2010) Genome-wide homozygosity signatures and childhood acute lymphoblastic leukemia risk. Blood 115 (22): 4472–4477.
Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E, Farrington S, Prendergast JG, Pittman AM, Theodoratou E, Smith CG, Olver B, Walther A, Barnetson RA, Churchman M, Jaeger EE, Penegar S, Barclay E, Martin L, Gorman M, Mager R, Johnstone E, Midgley R, Niittymaki I, Tuupanen S, Colley J, Idziaszczyk S, Thomas HJ, Lucassen AM, Evans DG, Maher ER, Maughan T, Dimas A, Dermitzakis E, Cazier JB, Aaltonen LA, Pharoah P, Kerr DJ, Carvajal-Carmona LG, Campbell H, Dunlop MG, Tomlinson IP (2010) Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 42 (11): 973–977.
Huhn S, Bevier M, Pardini B, Naccarati A, Vodickova L, Novotny J, Vodicka P, Hemminki K, Forsti A (2014) Colorectal cancer risk and patients' survival: influence of polymorphisms in genes somatically mutated in colorectal tumors. Cancer Causes Control 25 (6): 759–769.
Ishikawa Y, Ito S, Nagata K, Sakai LY, Bachinger HP (2016) Intracellular mechanisms of molecular recognition and sorting for transport of large extracellular matrix molecules. Proc Natl Acad Sci USA 113 (41): E6036–E6044.
Jiao S, Peters U, Berndt S, Brenner H, Butterbach K, Caan BJ, Carlson CS, Chan AT, Chang-Claude J, Chanock S, Curtis KR, Duggan D, Gong J, Harrison TA, Hayes RB, Henderson BE, Hoffmeister M, Kolonel LN, Le Marchand L, Potter JD, Rudolph A, Schoen RE, Seminara D, Slattery ML, White E, Hsu L (2014) Estimating the heritability of colorectal cancer. Hum Mol Genet 23 (14): 3898–3905.
Johns LE, Houlston RS (2001) A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol 96 (10): 2992–3003.
Kemp Z, Carvajal-Carmona L, Spain S, Barclay E, Gorman M, Martin L, Jaeger E, Brooks N, Bishop DT, Thomas H, Tomlinson I, Papaemmanuil E, Webb E, Sellick GS, Wood W, Evans G, Lucassen A, Maher ER, Houlston RS (2006) Evidence for a colorectal cancer susceptibility locus on chromosome 3q21-q24 from a high-density SNP genome-wide linkage scan. Hum Mol Genet 15 (19): 2903–2910.
Kerber RA, Neklason DW, Samowitz WS, Burt RW (2005) Frequency of familial colon cancer and hereditary nonpolyposis colorectal cancer (Lynch syndrome) in a large population database. Fam Cancer 4 (3): 239–244.
Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, Das J, Abyzov A, Balasubramanian S, Beal K, Chakravarty D, Challis D, Chen Y, Clarke D, Clarke L, Cunningham F, Evani US, Flicek P, Fragoza R, Garrison E, Gibbs R, Gumus ZH, Herrero J, Kitabayashi N, Kong Y, Lage K, Liluashvili V, Lipkin SM, MacArthur DG, Marth G, Muzny D, Pers TH, Ritchie GR, Rosenfeld JA, Sisu C, Wei X, Wilson M, Xue Y, Yu F, Dermitzakis ET, Yu H, Rubin MA, Tyler-Smith C, Gerstein M (2013) Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342 (6154): 1235587.
Kobayashi H, Komatsu S, Ichikawa D, Kawaguchi T, Hirajima S, Miyamae M, Okajima W, Ohashi T, Kosuga T, Konishi H, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Otsuji E (2015) Overexpression of denticleless E3 ubiquitin protein ligase homolog (DTL) is related to poor outcome in gastric carcinoma. Oncotarget 6 (34): 36615–36624.
Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61 (5): 1179–1188.
Lips EH, van Eijk R, de Graaf EJ, Oosting J, de Miranda NF, Karsten T, van de Velde CJ, Eilers PH, Tollenaar RA, van Wezel T, Morreau H (2008) Integrating chromosomal aberrations and gene expression profiles to dissect rectal tumorigenesis. BMC Cancer 8: 314.
Middeldorp A, Jagmohan-Changur SC, van der Klift HM, van Puijenbroek M, Houwing-Duistermaat JJ, Webb E, Houlston R, Tops C, Vasen HF, Devilee P, Morreau H, van Wezel T, Wijnen J (2010) Comprehensive genetic analysis of seven large families with mismatch repair proficient colorectal cancer. Genes Chromosomes Cancer 49 (6): 539–548.
Montazeri Z, Theodoratou E, Nyiraneza C, Timofeeva M, Chen W, Svinti V, Sivakumaran S, Gresham G, Cubitt L, Carvajal-Carmona L, Bertagnolli MM, Zauber AG, Tomlinson I, Farrington SM, Dunlop MG, Campbell H, Little J (2016) Systematic meta-analyses and field synopsis of genetic association studies in colorectal adenomas. Int J Epidemiol 45 (1): 186–205.
Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, Graff RE, Holst K, Moller S, Unger RH, McIntosh C, Nuttall E, Brandt I, Penney KL, Hartman M, Kraft P, Parmigiani G, Christensen K, Koskenvuo M, Holm NV, Heikkila K, Pukkala E, Skytthe A, Adami HO, Kaprio J (2016) Familial risk and heritability of cancer among twins in Nordic countries. JAMA 315 (1): 68–76.
Muzny D, Bainbridge M, Chang K, Dinh H, Drummond J, Fowler G, Kovar C, Lewis L, Morgan M, Newsham I, Reid J, Santibanez J, Shinbrot E, Trevino L, Wu Y, Wang M, Gunaratne P, Donehower L, Creighton C, Wheeler D, Gibbs R, Lawrence M, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander E, Gabriel S, Getz G, Ding L, Fulton R, Koboldt D, Wylie T, Walker J, Dooling D, Fulton L, Delehaunty K, Fronick C, Demeter R, Mardis E, Wilson R, Chu A, Chun H, Mungall A, Pleasance E, Robertson A, Stoll D, Balasundaram M, Birol I, Butterfield Y, Chuah E, Coope R, Dhalla N, Guin R, Hirst C, Hirst M, Holt R, Lee D, Li H, Mayo M, Moore R, Schein J, Slobodan J, Tam A, Thiessen N, Varhol R, Zeng T, Zhao Y, Jones S, Marra M, Bass A, Ramos A, Saksena G, Cherniack A, Schumacher S, Tabak B, Carter S, Pho N, Nguyen H, Onofrio R, Crenshaw A, Ardlie K, Beroukhim R, Winckler W, Getz G, Meyerson M, Protopopov A, Zhang J, Hadjipanayis A, Lee E, Xi R, Yang L, Ren X, Zhang H, Sathiamoorthy N, Shukla S, Chen P, Haseley P, Xiao Y, Lee S, Seidman J, Chin L, Park P, Kucherlapati R, Auman J, Hoadley K, Du Y, Wilkerson M, Shi Y, Liquori C, Meng S, Li L, Turman Y, Topal M, Tan D, Waring S, Buda E, Walsh J, Jones C, Mieczkowski P, Singh D, Wu J, Gulabani A, Dolina P, Bodenheimer T, Hoyle A, Simons J, Soloway M, Mose L, Jefferys S, Balu S, O'Connor B, Prins J, Chiang D, Hayes D, Perou C, Hinoue T, Weisenberger D, Maglinte D, Pan F, Berman B, Van Den Berg D, Shen H, Triche T Jr, Baylin S, Laird P, Getz G, Noble M, Voet D, Saksena G, Gehlenborg N, DiCara D, Zhang J, Zhang H, Wu C, Liu S, Shukla S, Lawrence M, Zhou L, Sivachenko A, Lin P, Stojanov P, Jing R, Park R, Nazaire M, Robinson J, Thorvaldsdottir H, Mesirov J, Park P, Chin L, Thorsson V, Reynolds S, Bernard B, Kreisberg R, Lin J, Iype L, Bressler R, Erkkilä T, Gundapuneni M, Liu Y, Norberg A, Robinson T, Yang D, Zhang W, Shmulevich I, de Ronde J, Schultz N, Cerami E, Ciriello G, Goldberg A, Gross B, Jacobsen A, Gao J, Kaczkowski B, Sinha R, Aksoy B, Antipin Y, Reva B, Shen R, Taylor B, Chan T, Ladanyi M, Sander C, Akbani R, Zhang N, Broom B, Casasent T, Unruh A, Wakefield C, Hamilton S, Cason R, Baggerly K, Weinstein J, Haussler D, Benz C, Stuart J, Benz S, Sanborn J, Vaske C, Zhu J, Szeto C, Scott G, Yau C, Ng S, Goldstein T, Ellrott K, Collisson E, Cozen A, Zerbino D, Wilks C, Craft B, Spellman P, Penny R, Shelton T, Hatfield M, Morris S, Yena P, Shelton C, Sherman M, Paulauskis J, Gastier-Foster J, Bowen J, Ramirez N, Black A, Pyatt R, Wise L, White P, Bertagnolli M, Brown J, Chan T, Chu G, Czerwinski C, Denstman F, Dhir R, Dörner A, Fuchs C, Guillem J, Iacocca M, Juhl H, Kaufman A, Kohl B 3rd., Van Le X, Mariano M, Medina E, Meyers M, Nash G, Paty P, Petrelli N, Rabeno B, Richards W, Solit D, Swanson P, Temple L, Tepper J, Thorp R, Vakiani E, Weiser M, Willis J, Witkin G, Zeng Z, Zinner M, Zornig C, Jensen M, Sfeir R, Kahn A, Chu A, Kothiyal P, Wang Z, Snyder E, Pontius J, Pihl T, Ayala B, Backus M, Walton J, Whitmore J, Baboud J, Berton D, Nicholls M, Srinivasan D, Raman R, Girshik S, Kigonya P, Alonso S, Sanbhadti R, Barletta S, Greene J, Pot D, Shaw K, Dillon L, Buetow K, Davidsen T, Demchok J, Eley G, Ferguson M, Fielding P, Schaefer C, Sheth M, Yang L, Guyer M, Ozenberger B, Palchik J, Peterson J, Sofia H, Thomson E (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487 (7407): 330–337.
Neklason DW, Kerber RA, Nilson DB, Anton-Culver H, Schwartz AG, Griffin CA, Lowery JT, Schildkraut JM, Evans JP, Tomlinson GE, Strong LC, Miller AR, Stopfer JE, Finkelstein DM, Nadkarni PM, Kasten CH, Mineau GP, Burt RW (2008) Common familial colorectal cancer linked to chromosome 7q31: a genome-wide analysis. Cancer Res 68 (21): 8993–8997.
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45 (2): 136–144.
Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC (2006) Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle 5 (22): 2676–2687.
Pardini B, Rosa F, Barone E, Di Gaetano C, Slyskova J, Novotny J, Levy M, Garritano S, Vodickova L, Buchler T, Gemignani F, Landi S, Vodicka P, Naccarati A (2013) Variation within 3'-UTRs of base excision repair genes and response to therapy in colorectal cancer patients: a potential modulation of microRNAs binding. Clin Cancer Res 19 (21): 6044–6056.
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29 (9): e45.
Saito K, Yamashiro K, Ichikawa Y, Erlmann P, Kontani K, Malhotra V, Katada T (2011) cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell 22 (13): 2301–2308.
Sasahira T, Kirita T, Nishiguchi Y, Kurihara M, Nakashima C, Bosserhoff AK, Kuniyasu H (2016) A comprehensive expression analysis of the MIA gene family in malignancies: MIA gene family members are novel, useful markers of esophageal, lung, and cervical squamous cell carcinoma. Oncotarget 7 (21): 31137–31152.
Sasahira T, Kirita T, Yamamoto K, Ueda N, Kurihara M, Matsushima S, Bhawal UK, Bosserhoff AK, Kuniyasu H (2014) Transport and Golgi organisation protein 1 is a novel tumour progressive factor in oral squamous cell carcinoma. Eur J Cancer 50 (12): 2142–2151.
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJ, Stinson J, Gnad F, Huntley MA, Degenhardt JD, Haverty PM, Bourgon R, Wang W, Koeppen H, Gentleman R, Starr TK, Zhang Z, Largaespada DA, Wu TD, de Sauvage FJ (2012) Recurrent R-spondin fusions in colon cancer. Nature 488 (7413): 660–664.
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314 (5797): 268–274.
Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, Stephens RM, Harris TJ, Munroe DJ, Wu X (2010) Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci USA 107 (7): 3001–3005.
Spain SL, Carvajal-Carmona LG, Howarth KM, Jones AM, Su Z, Cazier JB, Williams J, Aaltonen LA, Pharoah P, Kerr DJ, Cheadle J, Li L, Casey G, Vodicka P, Sieber O, Lipton L, Gibbs P, Martin NG, Montgomery GW, Young J, Baird PN, Morreau H, van Wezel T, Ruiz-Ponte C, Fernandez-Rozadilla C, Carracedo A, Castells A, Castellvi-Bel S, Dunlop M, Houlston RS, Tomlinson IP (2012) Refinement of the associations between risk of colorectal cancer and polymorphisms on chromosomes 1q41 and 12q13.13. Hum Mol Genet 21 (4): 934–946.
Spain SL, Cazier JB, Houlston R, Carvajal-Carmona L, Tomlinson I (2009) Colorectal cancer risk is not associated with increased levels of homozygosity in a population from the United Kingdom. Cancer Res 69 (18): 7422–7429.
Stolk RP, Rosmalen JG, Postma DS, de Boer RA, Navis G, Slaets JP, Ormel J, Wolffenbuttel BH (2008) Universal risk factors for multifactorial diseases: LifeLines: a three-generation population-based study. Eur J Epidemiol 23 (1): 67–74.
Sud A, Cooke R, Swerdlow AJ, Houlston RS (2015) Genome-wide homozygosity signature and risk of Hodgkin lymphoma. Sci Rep 5: 14315.
Theodoratou E, Montazeri Z, Hawken S, Allum GC, Gong J, Tait V, Kirac I, Tazari M, Farrington SM, Demarsh A, Zgaga L, Landry D, Benson HE, Read SH, Rudan I, Tenesa A, Dunlop MG, Campbell H, Little J (2012) Systematic meta-analyses and field synopsis of genetic association studies in colorectal cancer. J Natl Cancer Inst 104 (19): 1433–1457.
Timofeeva MN, Kinnersley B, Farrington SM, Whiffin N, Palles C, Svinti V, Lloyd A, Gorman M, Ooi LY, Hosking F, Barclay E, Zgaga L, Dobbins S, Martin L, Theodoratou E, Broderick P, Tenesa A, Smillie C, Grimes G, Hayward C, Campbell A, Porteous D, Deary IJ, Harris SE, Northwood EL, Barrett JH, Smith G, Wolf R, Forman D, Morreau H, Ruano D, Tops C, Wijnen J, Schrumpf M, Boot A, Vasen HF, Hes FJ, van Wezel T, Franke A, Lieb W, Schafmayer C, Hampe J, Buch S, Propping P, Hemminki K, Forsti A, Westers H, Hofstra R, Pinheiro M, Pinto C, Teixeira M, Ruiz-Ponte C, Fernandez-Rozadilla C, Carracedo A, Castells A, Castellvi-Bel S, Campbell H, Bishop DT, Tomlinson IP, Dunlop MG, Houlston RS (2015) Recurrent coding sequence variation explains only a small fraction of the genetic architecture of colorectal cancer. Sci Rep 5: 16286.
Tuohy TM, Rowe KG, Mineau GP, Pimentel R, Burt RW, Samadder NJ (2014) Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 120 (1): 35–42.
Ueki T, Nishidate T, Park JH, Lin ML, Shimo A, Hirata K, Nakamura Y, Katagiri T (2008) Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene 27 (43): 5672–5683.
van Eijk R, Stevens L, Morreau H, van Wezel T (2013) Assessment of a fully automated high-throughput DNA extraction method from formalin-fixed, paraffin-embedded tissue for KRAS, and BRAF somatic mutation analysis. Exp Mol Pathol 94 (1): 121–125.
van Roon EH, Boot A, Dihal AA, Ernst RF, van Wezel T, Morreau H, Boer JM (2013) BRAF mutation-specific promoter methylation of FOX genes in colorectal cancer. Clin Epigenet 5 (1): 2.
Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50 (1): 118–127.
Wiesner GL, Daley D, Lewis S, Ticknor C, Platzer P, Lutterbaugh J, MacMillen M, Baliner B, Willis J, Elston RC, Markowitz SD (2003) A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2-31.2. Proc Natl Acad Sci USA 100 (22): 12961–12965.
Wilson DG, Phamluong K, Li L, Sun M, Cao TC, Liu PS, Modrusan Z, Sandoval WN, Rangell L, Carano RA, Peterson AS, Solloway MJ (2011) Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J Cell Biol 193 (5): 935–951.
Young P, Ehler E, Gautel M (2001) Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol 154 (1): 123–136.
Zhang B, Jia WH, Matsuda K (2014) Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 46 (6): 533–542.
Acknowledgements
This publication is supported by COST Action BM1206 and MSMT LD14050. This study was funded by grants from the Dutch Digestive Foundation (MLDS FP13-13), Dutch Cancer Society (KWF UL2010-4656), Stichting Sasha Swarttouw-Hijmans and Leids Universiteits Fonds/ Gratama Stichting. This work was further supported by grants from Fondo de Investigación Sanitaria/FEDER (14/00173, 14/00230), Fundación Científica de la Asociación Española contra el Cáncer (GCB13131592CAST), Catalan Tumour Bank Network (Pla Director d’Oncologia, Generalitat de Catalunya), CERCA Programme (Generalitat de Catalunya) and Agència de Gestió d'Ajuts Universitaris i de Recerca (Generalitat de Catalunya, 2014SGR255). We are sincerely grateful to the Biobank of Hospital Clínic-IDIBAPS for technical help. The work was carried out in part at the Esther Koplowitz Centre and FPGMX. CIBERehd and CIBERER are funded by the Instituto de Salud Carlos III. We acknowledge core funding to the Wellcome Trust Centre for Human Genetics from the Wellcome Trust (090532/Z/09/Z) and the Oxford NIHR Biomedical Research Centre. We thank Rashmi Dibipsersat, Nesli Ozcelik, Melanie Schrumpf and Brendy van den Akker for their technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Schubert, S., Ruano, D., Elsayed, F. et al. Evidence for genetic association between chromosome 1q loci and predisposition to colorectal neoplasia. Br J Cancer 117, 1215–1223 (2017). https://doi.org/10.1038/bjc.2017.240
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.240
Keywords
This article is cited by
-
Mutational analysis of driver genes defines the colorectal adenoma: in situ carcinoma transition
Scientific Reports (2022)
-
Identification of oral squamous cell carcinoma markers MUC2 and SPRR1B downstream of TANGO
Journal of Cancer Research and Clinical Oncology (2021)
-
Moving the needle on colorectal cancer genetics: it takes more than two to TANGO
British Journal of Cancer (2018)
