
Abstract
Background:
Thousands of clinically relevant variations in BRCA1 and BRCA2 have been discovered and this poses a significant challenge with respect to the accurate detection, analysis turn-around time, characterisation and interpretation of these sequence variants.
Methods:
We evaluated the performance of different BRCA1/2 gene testing practices in routine diagnostic use in 20 European laboratories, with a focus on next-generation sequencing-based strategies as this is the technical approach implemented by or under adoption by most European clinical laboratories. Participant laboratories, selected on expertise and diagnostic service quality, tested 10 identical DNA samples containing a range of challenging pathogenic variants.
Results:
A small number of errors in the detection of pathogenic and significant variants were identified (2.6% diagnostic error rate). There was a high degree of concordance (>97%) across all laboratories for all variants detected. No systematic technical flaw was identified in the strategies employed across the participating laboratories.
Conclusions:
The discrepancies identified are most likely due to human error or the way the methodology has been implemented locally, for example, next-generation sequencing bioinformatics pipelines, rather than technical limitations of the methods. The choice of BRCA1/2 testing method will therefore depend on multiple factors including required throughput and turn-around times, access to equipment, expertise and budget.
Similar content being viewed by others


Main
Mutations in the BRCA1 and BRCA2 genes lead to an increased risk of developing breast or ovarian cancer. Women who are heterozygous for a BRCA1 or BRCA2 pathogenic variant have up to an 80% risk of developing breast cancer by age 90; and an ovarian cancer risk of about 55% with BRCA1 mutations and 25% with BRCA2 mutations (Malander et al, 2004; Majdak et al, 2005; Alsop et al, 2012; Dann et al, 2012). Thousands of clinically relevant variations in BRCA1 and BRCA2 have been discovered to date, and these are distributed widely throughout the entire coding regions and intron–exon boundaries (Casey, 1997). This poses a significant challenge with respect to the accurate variant detection, analysis turn-around time, characterisation and interpretation of BRCA1 and BRCA2 sequence variants.
BRCA1/2 mutation screening is provided by a large number of clinical diagnostic laboratories worldwide using various different analytical methods and technology platforms. Laboratory practice is changing rapidly due to increased demand for testing (Palma et al, 2008; Public Policy Institute A, 2015), the advent of treatment-focused genetic testing (Ledermann et al, 2014) and the rapid uptake of next-generation sequencing (NGS) technologies (Idris et al, 2013; Trujillano et al, 2015). It is critically important that the result of the BRCA1/2 test is accurate, as significant clinical decisions are being made based on the results, such as prophylactic surgery to reduce the risk of cancer development. Information on BRCA1/2 mutation status is now being used to inform treatment decisions such as the use of platinum-based chemotherapy (Christie et al, 2014) and to guide the use of PARP inhibitors (Ledermann et al, 2014).
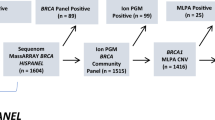
In this methodological assessment, we evaluated the performance of different BRCA1/2 gene testing methods in current routine clinical practice among European Molecular Genetics Quality Network (EMQN) members to determine how they compare at detecting clinically relevant BRCA1/2 variants. As a first step (Part A), the most common actively used methods for germline BRCA1/2 mutation analysis were determined through a questionnaire sent to active EMQN member laboratories participating in External Quality Assessment (EQA) for BRCA testing.
The study chose to focus on NGS and associated bioinformatics pipelines since this is the technical approach most labs are now using for BRCA1/2 testing. Therefore a subset of 20 laboratories representing 11 European countries were selected for further analytical evaluation in this study (Part B). Fifteen of these laboratories were using NGS, while the remaining five were using more traditional testing methodologies, that is, Sanger sequencing, denaturing high-performance liquid chromatography (dHPLC) and high-resolution melting HRM. Where possible, each method was undertaken by more than one participating laboratory to provide a comparison of the method rather than the individual laboratory. All of these laboratories were required to undertake BRCA1/2 testing on a range of highly characterised DNA samples with known BRCA1/2 genotypes, including a wide range of commonly encountered BRCA1 and BRCA2 variants encompassing a range of single-base substitutions, giving rise to missense, nonsense and splice-site mutations, insertion/deletion frameshift mutations, single exon deletions and large genomic rearrangements (LGR).
Materials and methods
Initial participant laboratory selection (Part A)
In Part A of the study a short survey of current BRCA1/2 gene screening methods was circulated to 156 members of the EMQN network participating in the 2014 EQA scheme for BRCA testing. The survey was used to establish the current state-of-the-art in BRCA testing among the EMQN member laboratories and to inform laboratory selection for Part B of the study.
Criteria for selection of laboratories for analytical sample investigation (Part B)
Laboratories were selected to participate in Part B of the study based on their responses to the Part A survey. Participation by selected laboratories was voluntary – all agreed to take part. These laboratories had demonstrated a significant diagnostic BRCA testing caseload per annum (>300 germline BRCA1/2 mutation analyses), conducted all testing in their own laboratory (no outsourcing or subcontracting of their testing process) and had demonstrated evidence of successful participation in three successive years in a recognised External Quality Assurance (EQA) scheme (e.g., EMQN, UK National External Quality Assessment Scheme (UK NEQAS) for Molecular Genetics, or College of American Pathology (CAP)). Where possible, we also selected laboratories which were accredited to the ISO15189 medical laboratory standard as this gave additional assurances and confidence that the lab was performing high-quality testing.
Laboratories using NGS as their primary mutation detection assay were sent an additional survey to establish the details of their bioinformatics analysis pipelines used to identify mutations, and the minimum standards of sequencing coverage that were applied to their diagnostic analysis.
Study procedure (Part C)
In total, 10 DNA samples extracted from lymphoblastoid cell lines as reference material (Table 1) were distributed to the 20 participating laboratories. These samples included eight specimens that contained a diverse range of BRCA1/2 mutation types (LGRs, missense, nonsense, splice and indels) while the remaining two contained no BRCA1/2 mutations. A total of seven pathogenic mutations and two variants of unknown significance (VUS) were supplied (see Table 1 for details). All participating laboratories were given two calendar months to complete their analyses using their standard clinical diagnostic methodology and were asked to report any clinically significant findings (i.e., the significant differences from the cDNA reference sequences BRCA1 NM_007294.3 and BRCA2 NM_000059.3) in addition to making available their ‘raw’ data if required. Large genomic rearrangements analysis of the samples was not mandated as a precondition for participation in the study. Laboratories were requested to analyse samples using their standard methodology with the intention that those using an NGS approach to detect LGRs would do so. Given this, some laboratories that used an NGS approach unsuitable for LGR analysis did not analyse the samples for LGRs although analysis with an independent method, for example, multiplex ligation-dependant probe amplification (MLPA) would be their standard practice.
Results
Laboratory distribution and BRCA1/2 screening methods
Eighty-nine (57%) laboratories responded to the survey. Twenty laboratories distributed across 11 EU countries were selected from the 89 applicants. In detail, six labs were from the UK, two labs each from France, Belgium, Germany and Italy, and one lab from Spain, Denmark, Greece, The Netherlands, Czech Republic and Austria, respectively. Fifteen of the participating labs used NGS as their primary BRCA1/2 mutation detection strategy, three used Sanger sequencing and two used a pre-screening strategy (high resolution melt (HRM) plus dHPLC analysis, or HRM analysis alone), followed by Sanger sequencing to define any variants identified via the pre-screening process. The NGS laboratories used a variety of different instruments and sequencing chemistries as well as sample preparation and library enrichment methods. The strategies used by the laboratories are described in more detail in Table 2.
Thirteen out of 20 (65%) of the participating laboratories analysed all the samples for LGRs. Eight of these used commercial MLPA probe sets while another five used NGS (with hybridisation enrichment) to directly detect LGRs. Three of the laboratories using MLPA only tested for the presence of BRCA1 deletions and duplications.
A wide range of bioinformatics approaches and tools were used with little consensus between the laboratories on the minimum and mean coverage cut-off parameters that were applied to NGS data. The target read depth across the region of interest varied from 30-fold to 500-fold coverage and the minimum acceptable read depth ranged from 10 × to 100 ×.
Concordance analysis of pathogenic BRCA1 and BRCA2 variants
All participant laboratories managed to complete their testing procedures for all of the samples provided. Given that there were nine pathogenic mutations/VUSs within the 10 samples, and allowing for the seven laboratories that did not analyse the samples for LGRs and three laboratories that only analysed for LGRs in BRCA1, there were 156 opportunities in total to detect pathogenic mutations/VUSs across the 20 laboratories (Table 3).
Importantly, no false-positive mutation/VUS calls were made across the 20 laboratories, resulting in 100% specificity of the mutation analyses. However, four pathogenic mutations were missed by three laboratories. The overall sensitivity of mutation/VUS detection across the laboratories was therefore 97.4% (152 out of 156).
In detail, laboratory 11 failed to identify two mutations; a 40 bp deletion in BRCA1 c.1175_1214del40, p.(Leu392Glnfs*5) and a splice-site mutation in BRCA2 c.7617+1G>T. Failure to identify these mutations is likely to be due to the specific bioinformatics analysis used in this centre, as laboratory 12 used exactly the same NGS technology and correctly identified both mutations.
One laboratory (laboratory 14) using the MRC-Holland MLPA BRCA1 P002-C3 kit missed a LGR. This same kit was also used by eight other laboratories to successfully detect BRCA1 LGRs in the study. Independent re-analysis of the MLPA data from this laboratory showed no evidence of the expected exon 3–8 duplication. After discussion with the laboratory we concluded that the most likely reason for failing to identify the mutation was a sample swap error during their MLPA analysis, as NGS analysis by the same laboratory did identify the rare BRCA1 c.4393A>C, p.(Ile1465Leu) VUS, which was also present in this sample.
Laboratory 16, using an HRM pre-screening strategy, failed to identify the BRCA2 nonsense mutation c.10G>T; p.(Gly4*) in one sample. Root cause analysis by the laboratory identified that the mutation was missed because the HRM profile was very similar to a known polymorphic variant within the amplicon containing the mutation. The laboratory did not undertake any confirmatory testing to directly identify the DNA change causing the aberrant melting profile.
Overall, the small number of errors in the detection of pathogenic and significant variants did not reveal any particular vulnerability in any of the mutation detection strategies employed across all participating laboratories.
Concordance analysis of all BRCA1/2 variants
In order to increase the power of the study to discriminate between the different analysis methods, we analysed the neutral (polymorphic) variants that were present in the test samples. Each participating laboratory was asked to report all the differences from the cDNA reference sequences (BRCA1 NM_007294.3 and BRCA2 NM_000059.3) that they identified in the sample set. We supplied a standard data sheet for completion in HGVS compliant format and also requested the zygosity of each variant detected. Since each laboratory had analysed intronic and non-coding regions of the samples to different extents we restricted the comparison to coding sequence variants only.
Across the 10 samples analysed there were 40 BRCA1 and 40 BRCA2 neutral coding region variants where the sequence in the test sample differed from the reference sequence. There were 20 unique single-nucleotide variants variants in total, 11 in BRCA1 and 9 in BRCA2 (Table 3). The analysis of the variant data was split into laboratories carrying out NGS analysis and those using Sanger sequencing. No analysis was done on the data from laboratories 15 and 16 as they were using a pre-screening only strategy with methods that do not specify the precise DNA sequence alteration. Notably, analysis of the pattern of variant genotypes reported by some laboratories showed that they had used an alternative reference sequence for BRCA2 (U43746.1) to analyse their data rather than the reference sequence, NM_000059.3, requested by the authors. Where the pattern of genotypes reported for a laboratory matched the use of U43746.1, the reported genotypes were scored against this reference sequence and the variances from expected were not counted as an error.
In total, of the 18 laboratories using a DNA sequencing technology (NGS or Sanger), 10 (55%) made no errors in the reported variant genotypes. Of the remaining 8 laboratories a total of 27 discordances from the expected genotype were reported. The discordances were broken down into four categories as follows: (i) Failure to detect a variant that deviates from the reference sequence (false negative) – 10 occurrences; (ii) Detection of a variant from the reference that is not present (false positive) – 6 occurrences; (iii) Zygosity incorrectly determined for a variant – 4 occurrences; (iv) Typographical error – 7 occurrences. Errors were categorised as typographical where a novel change was reported that shared at least three digits with a genuine variant that was not reported, for example, BRCA1 c.4873A>G heterozygous reported instead of BRCA1 c.4837A>G heterozygous.
Overall, the accuracy of genotyping the 1440 variants that were present in the 10 samples was 1413 out of 1440 (98.1%). This is close to the accuracy estimate of 97.4% from the 156 clinically relevant variants that were tested in the main phase of the study. The distribution of discordances across the sequencing laboratories is given in Table 4.
Interestingly both laboratories using an NGS screening technology that made errors in the reporting of clinically relevant variants also made errors in the reporting of all other variants (laboratories 11 and 14). There did not appear to be any overall pattern between the variants that were not detected between labs or samples, with errors seeming to be randomly distributed.
Interestingly, laboratory 4 conducted a more detailed analysis of the three errors that it had made in this part of the study. This laboratory had used Illumina sequencing chemistry coupled with a custom long PCR amplicon library preparation approach. All three errors (two for incorrect zygosity, and one for a variant not identified) were in the same sample (sample 6) and occurred in the same long PCR amplicon. One of the PCR primers for this amplicon overlaps the pathogenic BRCA2 deletion present in this sample (BRCA2 c.6842-?_7007+?del). Consequently, the presence of the large deletion leads to analytical interference preventing amplification of one of the BRCA2 alleles. Any mutation detection strategy dependant on PCR enrichment is prone to interference arising from such variants in the primer hybridisation sequences. Consequently, PCR primers for clinical assays should be carefully designed to avoid common variants that may cause interference in target amplification.
Discussion
In this study we have observed that various methods used by experienced clinical laboratories performed well on a range of challenging BRCA1/2 mutations. No single NGS method and associated bioinformatics pipeline was demonstrated to be superior, and were equally as capable of detecting the range of significant variants as more established methods. The results were shared between the study participants and have subsequently been used to improve practice in EQA schemes for BRCA testing and NGS.
The discrepancies in the identification of pathogenic and significant variants are most likely due to human error or the way the methodology has been implemented locally, for example, NGS bioinformatics pipelines, rather than technical limitations of the methods. Robust processes to eliminate sample mix-ups are essential and important to ensure high-quality testing. The extended analysis of neutral variants did not reveal any further specific vulnerabilities in the technologies used; however it did confirm the genotyping error rate of NGS analysis in BRCA1/2 to be in the range of 2–3%. This is consistent with the error rates identified in EQA schemes for BRCA1/2 (Mueller et al, 2004, Simon Patton, personal communication) and other inherited genetic disease (Dequeker et al, 2001; Seneca et al, 2008). The actual error rate in clinical practice is likely to be lower than the 2–3% identified in the study, as laboratories were not required to fully replicate their diagnostic reporting pathway. A significant proportion of the errors in the extended study were clerical, or likely to be, and the error would be expected to be detected during checks undertaken during the clinical reporting process. Consequently an error rate of 2–3% should be viewed as the upper boundary of diagnostic error in these experienced laboratories. Nevertheless the error rate in less experienced laboratories could be higher, as evidenced in EQA schemes for molecular pathology. In molecular testing for somatic mutations, the diagnostic testing process has evolved rapidly, as new treatment-related outcomes to gene alterations have become available (Normanno et al, 2013). Consequently, many laboratories have implemented testing strategies without rigorous validation or method verification. External Quality Assessment schemes in this area of diagnostic testing have demonstrated significantly higher error rates (Deans et al, 2011; Wong et al, 2012; Patton et al, 2014) but also improvements over time (Deans et al, 2013; Patton et al, 2014). It is therefore essential for all laboratories offering BRCA1/2 testing to carry out robust assay validation and participate in regular EQA in order to minimise the possibility of errors in diagnostic screening.
In conclusion, It is our understanding that the choice of BRCA1/2 testing method will therefore depend on multiple factors including required throughput and turn-around times, access to equipment, expertise and budget.
References
Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, Friedlander M, Fox S, Bowtell D, Mitchell G (2012) BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol 30: 2654–2663.
Casey G (1997) The BRCA1 and BRCA2 breast cancer genes. Curr Opin Oncol 9: 88–93.
Christie A, Cameron D, Gourley C (2014) Implications of BRCA1 and BRCA2 status for cancer clinical study outcomes. Clin Invest 4: 463–478.
Dann RB, DeLoia JA, Timms KM, Zorn KK, Potter J, Flake DD, Lanchbury JS, Krivak TC, Jemal A, Siegel R, Xu J, Ward E, Winter WE, Maxwell GL, Tian C, Carlson JW, Ozols RF, Rose PG, Ozols R, Bundy BN, Greer BE, Fowler JM, Clarke-Peraon D, Burger RA, Bookman MA, Brady MF, McGuire WP, Harper PG, Alberts DS, Friedlander M, Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Chen S, Iversen ES, Friebel T, Finkelstein D, Weber BL, Eisen A, Reedy M, Gallion H, Fowler JM, Kryscio R, Smith SA, Pal T, Wey JP, Betts JA, Krischer JP, Fiorca J, Arango H, Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, Chetrit A, Hirsch-Yechezkel G, Ben-David Y, Lubin F, Friedman, Sadetzki S, David YB, Chetrit A, Hirsch-Yechezkel G, Freidman E, Beck BD, Beller U, Thrall M, Gallion HH, Kryscio R, Kapali M, Armstrong DK, DeLoia JA, Weberpals J, Garbuio K, O’Brien A, Clark-Knowles K, Doucette S, Antoniouk O, Yang D, Khan S, Sun Y, Hess K, Shmulevich I, Sood AK, Weberpals JI, Clark-Knowles KV, Vanderhyden BC, Swisher EM, Gonzalez RM, Taniguchi T, Garcia RL, Walsh T, Goff BA, Venkitaraman AR, Zweemer RP, Shaw PA, Verheijen RM, Ryan A, Berchuck A, Ponder BA, Pike JM, Dorsman JC, Massuger LF, Ansink AC, Weegenaar J, Shvarts A, Russell PA, Pharoah PD, De Foy K, Ramus SJ, Symnads I, Wilson A, Tan DSP, Rothermundat C, Thomas K, Bancroft E, Eeles R, Shanley S, Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, Quinn JE, Carser JE, James CR, Kennedy RD, Harkin DP, Dhillon KK, Swisher EM, Taniguchi T, Chan KY, Ozcelik H, Cheung AN, Ngan HY, Khoo US, Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink N, Fong PC, Yap TA, Boss DS, Carden CP, Merhui-Roelvink M, Gourley C, Annunziata CM, Bates SE, Netork CGAR, Norquist BM, Garcia RL, Allison KH, Jokinen CH, Kernochan LE, Pizzi CC, Wilcox CB, Baysal BE, Gallion HH, Strange MA, DeLoia JA, Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D (2012) BRCA1/2 mutations and expression: response to platinum chemotherapy in patients with advanced stage epithelial ovarian cancer. Gynecol Oncol 125: 677–682.
Deans ZC, Bilbe N, O’Sullivan B, Lazarou LP, de Castro DG, Parry S, Dodson A, Taniere P, Clark C, Butler R (2013) Improvement in the quality of molecular analysis of EGFR in non-small-cell lung cancer detected by three rounds of external quality assessment. J Clin Pathol 66: 319–325.
Deans ZC, Tull J, Beighton G, Abbs S, Robinson DO, Butler R (2011) Molecular genetics external quality assessment pilot scheme for KRAS analysis in metastatic colorectal cancer. Genet Test Mol Biomarkers 15: 777–783.
Dequeker E, Ramsden S, Grody WW, Stenzel TT, Barton DE (2001) Quality control in molecular genetic testing. Nat Rev Genet 2: 717–723.
Idris SF, Ahmad SS, Scott MA, Vassiliou GS, Hadfield J (2013) The role of high-throughput technologies in clinical cancer genomics. Expert Rev Mol Diagn 13: 167–181.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, Barrett JC, Matulonis U (2014) Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15: 852–861.
Majdak EJ, De Bock GH, Brozek I, Perkowska M, Ochman K, Debniak J, Milczek T, Cornelisse CJ, Jassem J, Emerich J, Limon J, Devilee P, Xu CF, Chambers JA, Nicolai H, Mazoyer S, Puget N, Perrin-Vidonz L, Ozcelik H, Nedelcu R, Chan VWY, Liu HX, Cartegni L, Zhang MQ, Vega A, Campos B, de Pailleets BB, Feckenthal JD, Cartegni L, Krainer AR, Williams RS, Glover JN, Fleming MA, Potter JD, Ramirez CJ, Easton DF, Ford D, Bishop DT, Claus EB, Schildkraut JM, Thomposn WD, Culver HA, Cohen PF, Gldeea ME, Dorum A, Hovig E, Trope C, Geisler JP, Hatterman-Zogg MA, Rather JA, Gotlieb WH, Friedman E, Bar-Sade RB, Janezic SA, Ziogas A, Krumroy LM, Lu KH, Cramer DW, Muto MG, Modan B, Gak E, Sadebruchim RB, Moslehi R, Chu W, Karlan B, Mullen P, Miller WR, Mackay J, Sarantaus L, Vahteristo P, Bloom E, Stratton JF, Gayther SA, Russell P, Shephard JH, Boyd J, Sonoda Y, Federici MG, Górski B, Byrski T, Lubinski J, Perkowska M, Brożek I, Wysocka B, Boyd J, Rubin S, Johannsson OT, Tanstam J, Borg A, Lu KH, Broaddus RR, Lynch HT, Watson P, Bewtra C, Marcus JN, Watson P, Page DL, Narod SA, Sun P, Ghadirian P, Martinez MB, Ruan M, Fitzpatrick LA, Sparre LS, Kollind M, Carlstrom K, Ness RB, Cottreau C, Ness RB, Cottreau C, Kapplar J (2005) Prevalence and clinical correlations of BRCA1/BRCA2 unclassified variant carriers among unselected primary ovarian cancer cases–preliminary report. Eur J Cancer 41: 143–150.
Malander S, Ridderheim M, Måsbäck A, Loman N, Kristoffersson U, Olsson H, Nilbert M, Borg A, Pisani P, Parkin DM, Bray F, Ferlay J, Boyd J, Rubin SC, Kerber RA, Slattery ML, Ziogas A, Gildea M, Cohen P, Boyd J, Sonoda Y, Federici MG, Sekine M, Nagata H, Tsuji S, Piver MS, Rubin SC, Blackwood MA, Bandera C, Tavtigian SV, Simard J, Rommens J, Martin AM, Blackwood MA, Antin-Ozerkis D, Risch HA, McLaughlin JR, Cole DE, Antioniou A, Pharoah P, Narod S, Risch H, Venkitaraman AR, Koul A, Malander S, Loman N, Hakansson S, Johannsson O, Johansson U, Haraldsson K, Loman N, Zhang QX, Johannsson O, Olsson H, Borg A, Wagner T, Stoppa-Lyonnet D, Fleischmann E, Vallon-Christersson J, Cayanan C, Haraldsson K, Worley T, Vallon-Christersson J, Billack B, Borg A, Monteiro AN, Siddique H, Zou JP, Rao VN, Reddy ES, Futamura M, Arakawa H, Matsuda K, Takahashi H, Behbakht K, McGovern PE, Stratton JF, Gayther SA, Russell P, Berchuck A, Heron KA, Carney ME, Modan B, Hartge P, Hirsh-Yechezkel G, Takahashi H, Chiu HC, Bandera CA, Stratton JF, Thompson D, Bobrow L, Thompson D, Easton D, Rubin SC, Benjamin I, Behbakht K, David Y, Ben, Chetrit A, Hirsh-Yechezkel G, Zweemer RP, Verheijen RH, Coebergh JW, Johannsson OT, Ranstam J, Borg A, Olsson H, Werness BA, Ramus SJ, Whittemore AS (2004) One in 10 ovarian cancer patients carry germ line BRCA1 or BRCA2 mutations: results of a prospective study in Southern Sweden. Eur J Cancer 40: 422–428.
Mueller CR, Kristoffersson U, Stoppa-Lyonnet D (2004) External quality assessment for mutation detection in the BRCA1 and BRCA2 genes: EMQN’s experience of 3 years. Ann Oncol Off J Eur Soc Med Oncol 15 (Suppl 1): I14–I17.
Normanno N, Pinto C, Taddei G, Gambacorta M, Castiglione F, Barberis M, Clemente C, Marchetti A (2013) Results of the First Italian External Quality Assurance Scheme for somatic EGFR mutation testing in non-small-cell lung cancer. J Thorac Oncol 8: 773–778.
Palma MD, Domchek SM, Stopfer J, Erlichman J, Siegfried JD, Tigges-Cardwell J, Mason BA, Rebbeck TR, Nathanson KL (2008) The relative contribution of point mutations and genomic rearrangements in BRCA1 and BRCA2 in high-risk breast cancer families. Cancer Res 68: 7006–7014.
Patton S, Normanno N, Blackhall F, Murray S, Kerr KM, Dietel M, Filipits M, Benlloch S, Popat S, Stahel R, Thunnissen E (2014) Assessing standardization of molecular testing for non-small-cell lung cancer: results of a worldwide external quality assessment (EQA) scheme for EGFR mutation testing. Br J Cancer 111 (2): 413–420.
Public Policy Institute A (2015) Star Power: The Effect of Angelina Jolie’s Personal Story of BRCA1 Mutation on Testing Rates among Commercially Insured Women Insight on the Issues. Available at http://www.aarp.org/content/dam/aarp/ppi/2015-2/jolie-effect-on-brca-testing-aarp-insight.pdf.
Seneca S, Morris MA, Patton S, Elles R, Sequeiros J (2008) Experience and outcome of 3 years of a European EQA scheme for genetic testing of the spinocerebellar ataxias. Eur J Hum Genet 1629: 913–920.
Trujillano D, Weiss MER, Schneider J, Köster J, Papachristos EB, Saviouk V, Zakharkina T, Nahavandi N, Kovacevic L, Rolfs A (2015) Next-generation sequencing of the BRCA1 and BRCA2 genes for the genetic diagnostics of hereditary breast and/or ovarian cancer. J Mol Diagn 17: 162–170.
Wong NACS, Deans ZC, Ramsden SC (2012) The UK NEQAS for Molecular Genetics scheme for gastrointestinal stromal tumour: findings and recommendations following four rounds of circulation: Table 1. J Clin Pathol 65: 786–790.
Acknowledgements
We wish to express our gratitude to all participants for their hard work, prompt returns and their cooperation during this study:
Dr Angela Apessos, Genekor, Athens, Greece
Dr Christine Bell, Dept Medical Genetics, Aberdeen, United Kingdom
Dr Claude Houdayer, Service de Génétique, Institut Curie, Paris, France
Dr Dominique Vaur, Laboratoire de biologie et de génétique du cancer, Caen, France
Dr Erik Teugels, Free University of Brussels, Brussels, Belgium
Dr Eva Machackova, Masaryk Memorial Cancer Institute, Brno, Czech Republic
Dr Gabriele Wildhardt, bio.logis Center for Human Genetics, Frankfurt am Main, Germany
Dr Julian Nevado, Genomics of INGEMM-HULP, Madrid, Spain
Dr Marco Montagna, Hereditary Breast and Ovarian Cancer Unit, Padova, Italy
Dr Marjolijn Ligtenberg, Human genetics–Genome Diagnostics, Nijmegen, Netherlands
Dr Ruth Charlton, Yorkshire Regional Molecular Genetics Laboratory, Leeds, United Kingdom
Dr Stefania Tommasi, Istituto Tumori, Bari, Italy
Dr Stephen Abbs, East Anglia Medical Genetics Dept, Cambridge, United Kingdom
Dr Yvonne Wallis, West Midlands Regional Genetics Laboratory, Birmingham, United Kingdom
Mr Thomas van Overeem Hansen, Center for Genomic Medicine, Copenhagen, Denmark
Mrs Saskia Wehnelt, Myriad GmbH, Martinsried, Germany
Prof Gert Matthijs, Center for Human Genetics, Leuven, Belgium
Prof Klaus Wagner, Medical University of Graz, Graz, Austria
Prof Nazneen Rahman, TGLclinical, London, United Kingdom
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
GE and AK are employees of AstraZeneca plc. The remaining authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ellison, G., Wallace, A., Kohlmann, A. et al. A comparative study of germline BRCA1 and BRCA2 mutation screening methods in use in 20 European clinical diagnostic laboratories. Br J Cancer 117, 710–716 (2017). https://doi.org/10.1038/bjc.2017.223
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.223
Keywords
This article is cited by
-
The Landscape of BRCA Mutations among Egyptian Women with Breast Cancer
Oncology and Therapy (2023)
-
Reliable assessment of BRCA1 and BRCA2 germline variants by next-generation sequencing: a multicenter study
Breast Cancer (2021)
-
Biomarker testing in oncology – Requirements for organizing external quality assessment programs to improve the performance of laboratory testing: revision of an expert opinion paper on behalf of IQNPath ABSL
Virchows Archiv (2021)
-
Fertility preservation in BRCA mutation carriers—efficacy and safety issues: a review
Reproductive Biology and Endocrinology (2020)
-
Germline BRCA 1-2 status prediction through ovarian ultrasound images radiogenomics: a hypothesis generating study (PROBE study)
Scientific Reports (2020)
