
Abstract
Background:
Glioblastoma is the most common and most lethal primary brain cancer. CBF1 (also known as Recombination signal Binding Protein for immunoglobulin kappa J, RBPJ) is the cardinal transcriptional regulator of the Notch signalling network and has been shown to promote cancer stem-like cells (CSCs) in glioblastoma. Recent studies suggest that some of the malignant properties of CSCs are mediated through the activation of pro-invasive programme of epithelial-to-mesenchymal transition (EMT). Little is known whether CBF1 is involved in the EMT-like phenotype of glioma cells.
Methods:
In a collection of GBM neurosphere lines, we genetically inhibited CBF1 and investigated the consequences on EMT-related properties, including in vitro invasiveness by Boyden chambers assay, chemoresistance using a clinical drug library screen and glycolytic metabolism assessing live-cell extracellular acidification rate. We also compared CBF1 expression in cells exposed to low and high oxygen tension. In silico analysis in large-scale Western and Eastern patient cohorts investigated the clinical prognostic value of CBF1 expression in low- and high-grade glioma as well as medulloblastoma.
Results:
Mean CBF1 expression is significantly increased in isocitrate dehydrogenase 1 (IDH1) R132H mutant glioblastoma and serves as prognostic marker for prolonged overall survival in brain tumours, particularly after therapy with temozolomide. Hypoxic regions of glioblastoma have higher CBF1 activation and exposure to low oxygen can induce its expression in glioma cells in vitro. CBF1 inhibition blocks EMT activators such as zinc finger E-box-binding homeobox 1 (ZEB1) and significantly reduces cellular invasion and resistance to clinically approved anticancer drugs. Moreover, we indicate that CBF1 inhibition can impede cellular glycolysis.
Conclusions:
Mean CBF1 activation in bulk tumour samples serves as a clinical predictive biomarker in brain cancers but its intratumoral and intertumoral expression is highly heterogeneous. Microenvironmental changes such as hypoxia can stimulate the activation of CBF1 in glioblastoma. CBF1 blockade can suppress glioblastoma invasion in vitro in particular in cells undergone EMT such as those found in the hypoxic niche. Targeting CBF1 can be an effective anti-EMT therapy to impede invasive properties and chemosensitivity in those cells.
Similar content being viewed by others


Main
Metastasis and cell dissemination are the primary cancer-associated causes of death (Brabletz, 2012). In recent years, the paradigm of epithelial-to-mesenchymal transition (EMT), a multi-step molecular reprogramming process, has emerged as a potent driver of cancer dissemination and therapy resistance (Nieto et al, 2016). Consequently, tumour cell EMT is suggested to co-occur with the emergence of cancer stem cell (CSC) properties (Scheel and Weinberg, 2012). The identification and validation of potent target molecules that interfere with the EMT and CSC phenotype are of highest translational and clinical interest.
Glioblastoma is the most common and most aggressive primary malignant brain cancer characterised by highly infiltrative growth (Siegel et al, 2015). CBF1 (also known as Recombination signal Binding Protein for immunoglobulin kappa J, RBPJ) is the core transcriptional regulator of Notch signalling, an important phylogenetically conserved stem cell pathway known to have important roles in CSC in a variety of tumours, including glioblastoma (Fan et al, 2006; Wang et al, 2012). High levels of CBF1 have recently been identified as a mediator for chemoresistance of brain cancers against the anti-Notch drug class of γ-secretase inhibitors (GSIs) and targeting upstream or downstream targets of CBF1 can overcome this resistance. Blocking CBF1 in glioblastoma cells eradicates CSCs and reduces tumorigenicity (Xie et al, 2016). On the other hand, Notch signalling itself is a potent inducer of EMT in a variety of cancers (Timmerman et al, 2004) and activation of EMT promotes glioma cell invasion and therapy resistance (Kahlert et al, 2013; Siebzehnrubl et al, 2013). Given the suggested linkage of CSCs, Notch and EMT, we sought to investigate the effect of CBF1 inhibition on the mesenchymal transformation in brain cancer cells.
We show that targeting CBF1 leads to the impairment of a variety of cellular properties associated with EMT at least in part through the efficient suppression of EMT activators, including zinc finger E-box-binding protein 1 (ZEB1). Our observations show that inhibiting the recently identified CSC target impedes EMT-associated cellular processes in glioblastoma such as invasion and resistance against drugs other than GSIs. We also indicate that CBF1 is involved in regulating glioma cell glycolysis. Paradoxically, we reveal that CBF1 overall expression levels of bulk tumour samples are clinically positive prognostic in low- and high-grade adult and paediatric brain cancers both in Eastern and Western patient cohorts. Interestingly, the expression is significantly heterogeneous among different molecular subtypes of glioblastoma and medulloblastoma (MB). CBF1 becomes stimulated and alters its clinical prognostic value in response to alterations in the microenvironment such as induction of hypoxia or therapeutic treatment. CBF1 blockade is a potent therapeutic strategy that may be particular relevant against cells dependent on EMT.
Materials and methods
In silico analysis of clinical databases
The analysis of glioblastoma patient data was performed using three data sets: (1) public data for The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/); (2) public data from the French working group (Gravendeel et al, 2009); and (3) the member-restricted data of the Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn); were assessed. For TCGA, Affymetrix human exon 1.0 level 2 data was downloaded from https://gdc-portal.nci.nih.gov/. The French data are available through the R2 database (https://hgserver1.amc.nl). For the CGGA data, Illumina raw exon expression levels were used. The hereafter reported study using CGGA data was approved by the Beijing Tiantan Hospital institutional review board and tumour specimen quality control and determination of IDH1 mutation status has been described before (Yan et al, 2012). Long- and short-term overall survival of glioblastoma patients were analysed in the German Glioma Network data set (Reifenberger et al, 2014). Low-grade glioma (LGG) data were retrieved through CGGA, as well as from the French working group. MB results are based on the analysis of the raw data of the Pomeroy data set (Cho et al, 2011). Exon expression was combined using means to determine gene expression values and CBF1 high/low expression categories were assigned based on the final quartile for the TCGA and CGGA data and using a Scan method for the French data in R2. Survival analyses were calculated using the log-rank method for the TCGA and GCCA data and R2 for the French data.
RNA-seq. samples from the different anatomical regions of glioblastoma were obtained from the Ivy Glioblastoma Atlas Project from the Allen Institute for Brain Science downloaded in December 2016 (glioblastoma.alleninstitute.org). A total of 122 RNA samples were utilised in this analysis.
Cell culture, DNA constructs and viral infection
Glioblastoma neurosphere lines were generously provided by A. Vescovi, Milan, Italy (GBM1); MS Carro, Freiburg, Germany (BTSC407/407p) and G Riggins, Baltimore, MD, USA (JHH520/JHH). Cells were grown in serum-free suspension media as previously described (Kahlert et al, 2012). All cells are regularly characterised for mycoplasma negativity and cell line authenticity was assessed as previously described (Kahlert et al, 2016). Hypoxic cultures conditions were achieved by growing cells in an INVIVO2 200 hypoxic chamber (Baker and Baker Ruskinn, Sanford, MN, USA) under 2% oxygen for 48 h.
The following lentiviral constructs were used: empty control pLKO.1 TCR cloning vector (Addgene plasmid, Cambridge, MA, USA, #10878; Moffat et al, 2006) as well as pLKO.1 vector with cloned short hairpin RNA constructs directed against CBF1 mRNA as previously described (Asnaghi et al, 2014). Lentiviral particles were produced by transfecting 293T cells with the lentiviral packaging system as previously described (Kahlert et al, 2015) using GeneJuice (Merck Millipore, Darmstadt, Germany) per the manufacturer’s instructions. Supernatants collected 48 and 72 h after transfection were pooled and passed through a 0.45-micron filter before concentrated using polyethylene glycol and NaCl and then frozen at −80 °C until needed. Stable integration of pLKO.1 enables the serial selection of mutated cells through resistance against puromycin as previously described (Kahlert et al, 2015).
Stable integration of the reporter was selected through Neomycin resistance through serial passage (min. 7 days) in media supplemented with 1 mg ml−1 G418 (Carl Roth, Karlsruhe, Germany).
mRNA and protein analyses
The abundance of mRNA was assessed by conversion into complementary DNA and subsequent quantitative real-time PCR measurements using SYBR green-based fluorescence (Bio-Rad, Hercules, CA, USA). Relative quantification to housekeeping gene β-actin was assessed with ddCt-method. Primers were obtained through Sigma-Aldrich (St Louis, MO, USA), Germany and respective sequences can be found in Supplementary Figure S1. Western blotting was performed as described before (Kahlert et al, 2015); antibodies were used as per the manufacturer’s instructions (for specifications, see Supplementary Figure S1). Total protein abundance was determined colorimetrically using the DC Protein assay (Bio-Rad) and densitometry of the transferred proteins was performed electronically as previously described (Koch et al, 2016).
In vitro cell viability and invasion assay
Cellular growth of GBM neurospheres was assessed using the CellTiter Blue Viability assay (Promega, Madison, WI, USA) similarly as described before (Koch et al, 2016) plating 3000 cells per well in 100 μl growth media.
Assessment of cellular invasion was performed using modified 24-well Boyden Chamber assay similarly as described before (Kahlert et al, 2015).The inserts were coated with growth factor-reduced Matrigel (BD, Flanklin Lakes, NJ, USA) and incubated for 1 h at 37 °C. Subsequently, 75 000 cells suspended in 500 μl DMEM (Life Technologies, LTech, Carlsberg, CA, USA) were placed on top of each insert membrane. The bottom was filled with 700 μl DMEM media containing 10% foetal calf serum. All Boyden chamber assays were analysed 14 h after cell plating. The upper side of the membrane was then wiped carefully with a cotton swab to remove the rest of the plated cells. The membrane was then fixed in ice-cold methanol for 15 min and stained with Haematoxylin. The invasion of the cells was evaluated by counting the cell nuclei on the lower side of the membrane under a light microscope (five random high-power fields per insert).
Analysis of glycolysis
Determinants of glycolytic parameters in living cells were examined using Seahorse XFe96 extracellular flux analyser (Agilent Technologies, Santa Clara, CA, USA), which records extracellular acidification rates (ECARs) during oxidation of energy substrates. The assays were performed plating 25 000 cells per well on Laminin (Sigma-Aldrich) coated surface following the manufacturer’s instruction using glycolytic assay base medium (Agilent Technologies) and a settlement phase of 3 h. Individual port injections enabled controlled addition of glucose (final concentration: 10 mM), oligomycin A (final 1 μM) and 2-desoxyglucose (final 100 mM) according to the manufacturer’s description (‘glycolytic stress kit’). Data were analysed using the WAVE software (version 2.3., Agilent Technologies) to calculate the glycolytic parameters from the recorded ECAR values. A minimum of eight wells per group per repetition were plated and total protein content of each well was used for normalisation.
Drug cytotoxicity assays
Cell lines GBM1 and 407p with wild-type and inhibited CBF1 were tested for their sensitivity towards a collection of clinically approved drugs by automated screening. Inhibitors were tested in 384-well plates in 9 dilution steps with a concentration range from 0.005 to 25 μM using D300e Digital Dispenser (Tecan, Zurich, Switzerland). The cell suspension was dispensed in neurosphere media at the appropriate cell concentration per well with a volume of 30 μl per well using Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, Waltham, MA, USA) followed by incubation for 72 h. We predefined the optimal cell number of 3000 cells per well for all tested cell lines by using pLKO.1 control cells. Viability readout was carried out by adding 30 μl of the CellTiter Glo Reagent (1:1 diluted with PBS) into the wells, shaking the plate at about 900 r.p.m. for 2 min and incubating the plates for 10 min in the dark followed by luminescence quantification using Spark 10M multimode plate reader (Tecan).
Targeted drug assays with selected drugs were performed with Titer Blue assay similarly as described above.
Statistical analyses
Significance was tested with Student’s t-test performed in Statistica v5.1. (Statsoft, Tulsa, OK, USA). Bar plots present the mean and s.e.m.
Results
Inhibition of CBF1 results in notch-like gene signature
We created suspension cell models of suppressed CBF1 expression using RNA interference technology. Figure 1A shows the verification of CBF1 blockade by western blotting. Knockdown efficiency of ⩾70% (except for 407p shCBF1a construct) was achieved. CBF1 has key activating and repressing roles on Notch signalling depending on the baseline pathway activation level (Bray, 2006). Assessing Notch pathway target gene activation, we observed a significant induction of HES1, HEY1 and HES5 after CBF1 suppression (Figure 1B). Of note, GBM1 and 407p are well characterised concerning their Notch dependency and GSI treatment leads to the reduction of HES and HEY expression (Fan et al, 2006; Kahlert et al, 2016).

CBF1 knockdown results in expression of Notch-like signature. (A) CBF1 knockdown efficiency as assessed by western blotting-based protein quantification. (B) Cells with blocked CBF1 show increased levels of Notch pathway target gene expression (HES1, HES5 and HEY1) as compared with control cells carrying empty pLKO.1 vector. *P=0.05, **P=0.01, ***P=0.001.
CBF1 blockade impairs cellular invasion and reduces the expression of activators of EMT
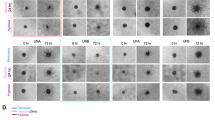
A main hallmark of EMT is the induction of cellular invasiveness leading to increased tumour cell dispersion (Brabletz, 2012). We found a striking impairment of invasive potential upon CBF1 blockade as compared with control cells in all tested cell lines (Figure 2A). We and others have described the key role of EMT activator ZEB1 in promoting glioblastoma cell invasion (Kahlert et al, 2012, 2015; Siebzehnrubl et al, 2013; Koch et al, 2016). Strikingly, ZEB1 was strongly reduced when blocking CBF1 (Figure 2B). Moreover, we tested the transcription levels of a collection of other known EMT markers and identified CD44s (standard isoform) and SNAI1 to be reduced when blocking CBF1 (Figure 2C). No effect on cellular growth was observed in any genetic condition (Figure 2D).

Inhibition of CBF1 impairs cellular invasion of glioma neurospheres. (A) GBM neurospheres with inhibited CBF1 are significantly less invasive in vitro as shown by representative microscope capturing and quantification of Boyden chamber assays. (B) CBF1 inhibition robustly suppresses the expression of pro-invasive EMT-activator ZEB1 as shown by western blotting. (C) Additional qPCR analysis revealed CBF1 blockade-suppressed CD44s and SNAI1 transcription. (D) CBF1 inhibition did not alter the growth of GBM neurospheres as tested by Titer Blue assay. *P=0.05, **P=0.01, ***P=0.001. A full colour version of this figure is available at the British Journal of Cancer journal online.
Hypoxia contributes to intratumoral heterogeneity of CBF1 expression
To characterise the clinical importance of CBF1, we comprehensively assessed its predictive value in various clinical data sets. Interestingly, we found that its expression is predominantly high in pseudopalisades (P⩾0.001) and peri-necrotic regions (Figure 3A), areas known to be severely hypoxic and containing a population of high invasive cells (Brat et al, 2004). Low oxygen tension is a potent stem cell niche factor (Mohyeldin et al, 2010) and a fundamental driver of cellular invasiveness through induction of EMT (Jiang et al, 2011; Kahlert et al, 2015). Interestingly, we found several consensus core hypoxic response elements (HRE) as defined by RCGTG (Wenger et al, 2005) in the promoter region of CBF1. Using the eukaryotic promoter database (http://epd.vital-it.ch), we identify n=3 HRE in the CBF1 core promoter (500 bp upstream of transcriptional start site) and n=8 HRE in the extended promoter region (=1500 bp upstream TSS). In concordance, exposure of cells to hypoxia, molecularly verified by the induction of hypoxia inducible factor 1α (HIF1α), can induce the expression of CBF1 as shown for GBM1 and strongly in 407p (Figure 3B). No CBF1 induction was noticed for JHH. We also noticed that CBF1 blockade can suppress HIF1α in normoxia, suggesting a potential reciprocal HIF1α/CBF1 loop (Supplementary Figure S2).

CBF1 is increased in hypoxia. (A) CBF1 mRNA is increased in hypoxic areas of glioblastoma samples as defined as pseudopalisades (**P=0.01, ***P=0.001) and peri-necrotic areas retrieved from the IVY Glioblastoma data set. (B) Hypoxic exposure (H, 2% oxygen, 48 h) increased the levels of HIF1α and led to induction of CBF1 (GBM1 and 407p) as compared with cells grown in normoxia (N). A full colour version of this figure is available at the British Journal of Cancer journal online.
Taken together, our data indicate that CBF1 in glioblastoma is intratumoral heterogeneously expressed and can be influenced by altered tumour microenvironment, such as induction of hypoxia.
CBF1 inhibition increases sensitivity to inhibitors of epidermal growth factor receptor (EGF-R), vascular endothelial growth factor receptor (VEGF-R) and m-TOR
A cardinal feature of mesenchymal-transformed cancer cells is a high level of resistance to therapeutic interventions (Singh and Settleman, 2010). Testing cell lines GBM1 and 407p with and without blocked CBF1, we identified drugs belonging to the classes of inhibitors of EGF-R (=AZD-9192), VEGF-R (=Cediranib) and m-TOR (PF-04691502) to act more potent when CBF1 is inhibited (Figure 4). More follow-up experiments are needed to exactly decipher the underlying mechanisms of this observation. Of note, no decrease of resistance to standard-of-care chemotherapeutic temozolomide (TMZ) was noticed when blocking CBF1. This was true for glioblastoma cell lines with high and low TMZ resistance (Supplementary Figure S3).

CBF1 inhibition reduces chemoresistance to members of various drug classes. Glioma cells with blocked CBF1 are more sensitive to the treatment with EGF-R inhibitor AZD 9291 (mesylate), VEGF-R inhibitor Cediranib and PI3K/mTOR inhibitor PF-04691502 as compared with pLKO.1 control cells. (A) Toxicity curves represent the results of the automated drug library screen, whereas (B) shows bar blots of targeted drug tests normalised to corresponding isogenic cells treated with drug solvent only. *P=0.05, **P=0.01.
CBF1 blockade can reduce cellular glycolysis
EMT-promoted tumour cell invasion has been associated with the preferred use of glycolysis as primary energy source (Shiraishi et al, 2014). Therefore, we investigated whether CBF1 blockade has any effects on glycolytic metabolism by quantifying ECAR. Although in different response values, all tested cell lines showed significantly reduced ECAR when CBF1 is inhibited (Figure 5A). Our observation was supported by effectively suppressed transcript levels of genes involved in glycolytic performance at least in two of the three tested cell lines: Cells with blocked CBF1 showed suppressed mRNA levels of enolase 1 (all tested cell lines) and pyruvate kinase M2 (GBM1, 407p) as well as hexokinase 1 (GBM1) and hexokinase 2 (GBM1, 407p) (P=0.05, Figure 5B). In JHH, CBF1 suppression did not affect the transcription of the analysed genes. Further studies are needed to comprehensively assess the underlying mechanisms of CBF1 inhibition on glycolysis and other metabolic networks in glioblastoma cells.

CBF1 inhibition can reduce glycolytic metabolism. (A) Cells with blocked CBF1 (=shRNAi construct b) have significantly reduced glycolytic activity as assessed by quantification of ECAR under defined glucose supply. (B) CBF1 knockdown can reduce the expression levels of central genes of glycolysis (enolase1=ENO1; hexokinase 1/2=HK1/2; and pyruvate kinase isoenzyme M2=PKM2) at various degrees with the strongest inhibition observed in GBM1 (all tested genes) to strong in 407p (ENO1 and PKM2) to moderate in JHH (ENO1). Interestingly, levels of gene inhibition correlates with the percentage of decreased ECAR from the highest in GBM1 to high in 407p to moderate in JHH. *P=0.05, ***P=0.001.
High CBF1 expression is a positive prognostic factor for overall survival
Depending on the activation level of Notch signalling, CBF1 acts as a transcriptional pathway activator or suppressor (Bray, 2006). Given its bivalent nature, the role of CBF1 during tumorigenesis is controversially discussed. Reports on clinical data mostly focussed on the tumour expression levels compared with tissue matching healthy counterparts but little is known about the prognostic relevance of CBF1 with regards to overall survival.
Comparing the overall expression levels of various brain tumour types separated by WHO grade, glioblastomas have relatively reduced CBF1 expression (Figure 6A). Survival analysis of two independent data sets of Western patients (French and TCGA) reveals patients with high CBF1 survive significantly longer. Assessing the German Glioma Network, we did not find any significant differences in CBF1 levels between long- or short-term survivors (data not shown) nor did we find a survival difference in dependency of CBF1 activation in the Chinese cohort (Figure 6B). Interestingly, the survival benefit of high CBF1 is of particular eminent in female glioblastoma patients. Moreover, it also becomes strongly significant when the patients were treated with standard-of-care adjuvant chemotherapy TMZ (Figure 6C), but no change in mean mRNA expression levels in treated and untreated patients are noticed (data not shown). CBF1 levels are significantly increased in proneural glioblastoma as compared with any other molecular subgroup as divided after Verhaak et al (2010) (Figure 6E). Only in proneural glioblastoma, CBF1 mRNA levels have clinical predictive value (Supplementary Figure S4). Of note, glioblastoma with mutant IDH1 have strongly significantly increased CBF1 expression as tumours with wild-type IDH1 both in the TCGA and CGGA data sets. Survival analysis reveals that IDH1 mutation status stratifies into clinically significant different groups only in tumours with high CBF1 expression level and has no significance in tumours with low CBF1 activation (Figure 6F).

CBF1 expression in brain tumours is clinically prognostic and intertumoral heterogeneous. (A) Mean CBF1 expression in different LGGs and glioblastoma showing relative low level of activation in the latter. PA= pilocytic astrocytoma; A=astrocytoma; OA=oligoastrocytoma; OD=oligodendroglioma; GBM=glioblastoma. (B) High levels of CBF1 predicts prolonged overall survival in glioblastoma patients from the Western populations (data sets: French, TCGA) but not in the Eastern cohort (CGGA). (C) The prognostic value is particularly eminent in female patients and (D) after therapy with alkylating agent TMZ. (E) CBF1 is strongly increased in the proneural subgroup of glioblastoma (F) as well as in tumours with R132H mutant IDH1. The predictive value of IDH1 mutation status with regards to overall survival does apply to tumours with high CBF1 expression but not under low CBF1 activation. *P=0.05, **P=0.01, ***P=0.001.
In addition, we analysed the clinical relevance of CBF1 levels in other types of brain cancer. Similar to GBM, Figure 7A shows that high CBF1 predicts for significantly longer survival for patients with LGG (Figure 7A) and MB (Figure 7B). Interestingly, comparing the molecular subgroups of MB, Group 3, the subset with the worst clinical outcome, has relatively low overall CBF1 activation. Paradoxically, only in this subtype high CBF1 is associated with significant shortened overall survival. In summary, analysing various clinical databases we identified significant heterogeneous activation levels of CBF1 in one cancer type both in adult and paediatric brain tumours. Our multi-cancer analysis suggests that elevated mean CBF1 mRNA expression derived from bulk tumour specimens predict for prolonged overall survival.

Clinical prognosis of CBF1 expression in LGGs and medulloblastomas. (A) High CBF1 expression correlates with very significantly prolonged survival of patients with LGG (WHO II+III). (B) Intertumoral heterogeneous activation of CBF1 in medulloblastoma subtypes with clinical worst prognostic Group 3 tumours having the lowest CBF1 level. High CBF1 predicts for significant longer survival in all medulloblastoma (and SHH subgroup) but predicts significantly shortened overall survival in Group 3 and Group 4 tumours. *P=0.05, **P=0.01, ***P=0.001.
Discussion
Targeting the Notch signalling pathway is an efficient strategy to eradicate CSCs with several promising clinical trials underway (Kahlert et al, 2017). CBF1 is a central signal mediator of the Notch network that has both pathway-activating and -suppressive functions (Bray, 2006). Our in vitro data with glioblastoma neurospheres implies that CBF1 blockade leads to activation of Notch as evidenced by robust induction of pathway target gene expression. We and others have reproducibly shown that HES1/5 and HEY1 transcript levels are robust reporters to determine Notch pathway activation status (Fan et al, 2006; Kahlert et al, 2016). However, we cannot exclude that in our cell models those selected genes are dysregulated by other factors than Notch in response to CBF1 inhibition. As such, Notch target genes are also influenced by other factors, which include androgen (Chen et al, 2010), c-jun N-terminal protein kinase (Curry et al, 2006) or Hedgehog (Benito-Gonzalez and Doetzlhofer, 2014) signalling. Importantly, CBF1 blockade can also indirectly induce the expression of CBF1-regulated genes by epigenetic activation of the promoter region of target genes via histone modification (Kulic et al, 2015). Therefore, such Notch-independent cues acting on CBF1-sensitive genes may be an alternative explanation for the observed induction of target genes after CBF1 suppression.
Studied in a variety of cancers, CBF1 have been described to possess both proto-oncogenic and tumour-suppressive functions (Bild et al, 2006; Kulic et al, 2015; Lv et al, 2015; Procopio et al, 2015). In our clinical assessments, overall high CBF1 expression correlates with favourable clinical prognosis in LGG and high-grade glioma as well as in MB. Contrary, CBF1 was recently discovered to serve as a potent target to diminish tumorigenicity and chemoresistance in glioblastoma (Xie et al, 2016). The authors also found high CBF1 expression to correlate with shortened overall survival. Of note, they restricted their analysis to glioblastoma without CpG island methylator phenotype (non-G-CIMP), the most aggressive epigenetic subtype. Interestingly, also in the most aggressive molecular subtypes of MB (Groups 3 and 4) we found high CBF1 to predict shortened survival. We suggest that CBF1 expression in brain cancers is intratumoral and intertumoral heterogeneous and its function is dependent on the genetic and (micro) environmental context. Of note, it was revealed that gene expression profiles in gliomas are highly heterogeneous and one tumour is comprised of areas belonging to distinct molecular subgroups (Morrissy et al, 2017). Therefore, gene expression data derived from one bulk sample does not resemble the complex genomic of the tumour and activation of CBF1 may differ significantly depending on spatial distribution and mutational background. Defining the mutation status of IDH1was recently included in the WHO guidelines for clinical diagnosis of brain cancers (Louis et al, 2016). We found that subclassification according to IDH1 mutation status also significantly separates tumours in high vs low CBF1 expression. IDH1 mutations frequently occur in secondary glioblastoma or diffusely growing LGGs, whereas primary de novo glioblastoma rarely show mutations in the IDH1 gene. Of note, the survival benefit of IDH1 mutation remains only significant in tumours with high CBF1 expression. CBF1 activation is also only prognostic in patients who received TMZ. The clinical data on IDH1 status and TMZ therapy suggests that CBF1 is increased in secondary glioblastoma and indicates that its function may be influenced by microenvironmental stress factors. This is further supported through the observed intratumoral heterogeneity of CBF1 expression in glioblastoma with elevated levels in pseudopalisades and peri-necrotic foci, both regions of severe hypoxia (Brat et al, 2004). Evidenced by in silico and in vitro data, we show that at least in a subset of glioblastoma CBF1 is induced by hypoxia. We detect multiple HRE in the CBF1 promoter region and observed that the exposure to low oxygen tension elevates CBF1 protein levels in glioma neurospheres.
Hypoxia is a well-known niche for stem cells in development and disease (Mohyeldin et al, 2010). In concordance, recent work has discovered that targeting CBF1 or its downstream mediators effectively diminishes glioma stem-like cells, especially those that are resistant to anti-Notch pathway-directed therapies (Xie et al, 2016). However, little is known about the effect of targeting CBF1 on tumour cell dissemination and invasion. As EMT has been revealed as a main mediator of glial cancer cell invasion (Kahlert et al, 2013; Siebzehnrubl et al, 2013), we sought to study the consequences of inhibiting CBF1 on pro-invasive mesenchymal transformation. Strikingly, blocking CBF1 led to strong reduction of invasive behaviour in vitro accompanied with a reduction of EMT-activator ZEB1 in all tested cell lines. Notch signalling is well described to promote cell invasion through induction of EMT in a variety of solid cancers (Brabletz et al, 2011). Our data now extend to glioma cell invasion to be regulated through the multivalent axis of CBF1/EMT signalling and suggests that a central member of the Notch pathway is involved in EMT-like processes in glioblastoma. Of note, RNAi construct a only minimally suppresses ZEB1 in 407p cells but still results in strong significant impairment of invasion. Besides showing that also other EMT promoters are suppressed in CBF1 knockdown cells, we cannot exclude the induction of other genetic responses that suppress cell invasiveness in response to modulating CBF1 expression.
CBF1/Notch1–ZEB1 interplay was recently described in lung cancer to regulate resistance against inhibitors of EGF-R signalling (Zhang et al, 2016). Our data extend the relevance of CBF1/EMT axis regulating chemosensitivity to cells of glial cancers as CBF1 blockade in glioma neurospheres can increase the cytotoxicity of inhibitors of EGF-R, VEGF-R or PI3K/mTOR signalling.
With the technical advantages to study cellular energetic networks, the influence of oncoproteins on metabolism has become increasingly apparent. Recent developments in cancer research led to the consideration that targeting metabolic networks can be a promising way to effectively target cancer malignancy, including in glioblastoma (Kahlert et al, 2017). In breast cancer, EMT induces glycolytic metabolism (Kondaveeti et al, 2015) and glycolysis is the primary energy pathway used for breast and prostate cancer cell motility (Shiraishi et al, 2015). Also the ‘go or grow’ potential of glioblastoma cells is regulated by active metabolic switch featuring glycolytic activation in regions of hypoxia leading to high invasiveness and low proliferation (Kathagen et al, 2013). Our data now indicate that targeting the CBF1/EMT axis in glioblastoma cells can cause an inhibition of glycolysis, which may be at least in part mediated through suppression of cardinal enzymatic regulators of glucose breakdown, such as hexokinase 1/2, enolase 1 or pyruvate dehydrogenase M2. This is particular exciting as glycolytic enzymes are major targets in cancer therapy with the development of promising clinical inhibitors underway (Scatena et al, 2008). Of note, PKM and HK are HIF transcription factor target genes, and given the fact that CBF1 can inhibit HIF1α, our data cannot exclude that the glycolytic effect is caused indirectly. Although we did not see noticeable expression levels of HIF2α under normoxia (data not shown), we also cannot exclude that changes in the activation of cardinal glioma-oncogene HIF2α (Li et al, 2009) contribute to the observed phenotype upon CBF1 blockade. Of note, we recently also identify ZEB1 as a potent metabolic regulator in glioma cells but did not on investigating its effects on glycolysis (Koch et al, 2016). In addition, we see variations in the degree of inhibition of glycolysis and its related genes among our tested cell models, indicating that the influence of CBF1 on glucose metabolism strongly varies among different tumours.
In summary, we provide evidences that blocking CBF1 in glioblastoma cells can suppress cellular invasion and chemoresistance against multiple drug classes as least partly through inhibiting activators of EMT, including ZEB1. We show that CBF1 can be induced by environmental stimuli and its activation is intratumoral and intertumoral heterogeneous. CBF1 inhibition is a potent therapeutic target to block invasion and therapy resistance, which may be particularly relevant to fight hypoxic-driven EMT cells.
Change history
27 June 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Asnaghi L, Lin MH, Lim KS, Lim KJ, Tripathy A, Wendeborn M, Merbs SL, Handa JT, Sodhi A, Bar EE, Eberhart CG (2014) Hypoxia promotes uveal melanoma invasion through enhanced Notch and MAPK activation. PLoS One 9: e105372.
Benito-Gonzalez A, Doetzlhofer A (2014) Hey1 and Hey2 control the spatial and temporal pattern of mammalian auditory hair cell differentiation downstream of hedgehog signaling. J Neurosci 34: 12865–12876.
Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi M-B, Harpole D, Lancaster JM, Berchuck A, Olson JA, Marks JR, Dressman HK, West M, Nevins JR (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439: 353–357.
Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J, Brabletz T (2011) The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J 30: 770–782.
Brabletz T (2012) To differentiate or not—routes towards metastasis. Nat Rev Cancer 12: 425–436.
Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Meir EGV (2004) Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 64: 920–927.
Bray SJ (2006) Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7: 678–689.
Chen H, Libertini SJ, George M, Dandekar S, Tepper CG, Al-Bataina B, Kung H-J, Ghosh PM, Mudryj M (2010) Genome-wide analysis of androgen receptor binding and gene regulation in two CWR22-derived prostate cancer cell lines. Endocr Relat Cancer 17: 857–873.
Cho Y-J, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, Gajjar A, Delattre O, Kool M, Ligon K, Meyerson M, Mesirov JP, Pomeroy SL (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29: 1424–1430.
Curry CL, Reed LL, Nickoloff BJ, Miele L, Foreman KE (2006) Notch-independent regulation of Hes-1 expression by c-Jun N-terminal kinase signaling in human endothelial cells. Lab Invest 86: 842–852.
Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li Y-M, Eberhart CG (2006) Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66: 7445–7452.
Gravendeel LAM, Kouwenhoven MCM, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LBC, Kloosterhof NK, De Moor B, Eilers PHC, van der Spek PJ, Kros JM, Sillevis Smitt PAE, van den Bent MJ, French PJ (2009) Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res 69: 9065–9072.
Jiang J, Tang Y, Liang X (2011) EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther 11: 714–723.
Kahlert UD, Cheng M, Koch K, Marchionni L, Fan X, Raabe EH, Maciaczyk J, Glunde K, Eberhart CG (2016) Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int J Cancer 138: 1246–1255.
Kahlert UD, Maciaczyk D, Doostkam S, Orr BA, Simons B, Bogiel T, Reithmeier T, Prinz M, Schubert J, Niedermann G, Brabletz T, Eberhart CG, Nikkhah G, Maciaczyk J (2012) Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett 325: 42–53.
Kahlert UD, Mooney SM, Natsumeda M, Steiger H-J, Maciaczyk J (2017) Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. Int J Cancer 140: 10–22.
Kahlert UD, Nikkhah G, Maciaczyk J (2013) Epithelial-to-mesenchymal(-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett 331: 131–138.
Kahlert UD, Suwala AK, Raabe EH, Siebzehnrubl FA, Suarez MJ, Orr BA, Bar EE, Maciaczyk J, Eberhart CG (2015) ZEB1 promotes invasion in human fetal neural stem cells and hypoxic glioma neurospheres. Brain Pathol 25: 724–732.
Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, Günther HS, Soriano R, Modrusan Z, Sandmann T, Kuhl C, Tissier A, Holz M, Krawinkel LA, Glatzel M, Westphal M, Lamszus K (2013) Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol (Berl) 126: 763–780.
Koch K, Hartmann R, Schröter F, Suwala AK, Maciaczyk D, Krüger AC, Willbold D, Kahlert UD, Maciaczyk J, Koch K, Hartmann R, Schröter F, Kora Suwala A, Maciaczyk D, Caroline Krüger A, Willbold D, Dietrich Kahlert U, Maciaczyk J (2016) Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 7: 73414–76431.
Kondaveeti Y, Guttilla Reed IK, White BA (2015) Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett 364: 44–58.
Kulic I, Robertson G, Chang L, Baker JHE, Lockwood WW, Mok W, Fuller M, Fournier M, Wong N, Chou V, Robinson MD, Chun H-J, Gilks B, Kempkes B, Thomson TA, Hirst M, Minchinton AI, Lam WL, Jones S, Marra M, Karsan A (2015) Loss of the Notch effector RBPJ promotes tumorigenesis. J Exp Med 212: 37–52.
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN (2009) Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15: 501–513.
Louis DN, Perry A, Reifenberger G, Deimling A, von Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (Berl) 131: 803–820.
Lv Q, Shen R, Wang J (2015) RBPJ inhibition impairs the growth of lung cancer. Tumour Biol 36: 3751–3756.
Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124: 1283–1298.
Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A (2010) Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7: 150–161.
Morrissy AS, Cavalli FMG, Remke M, Ramaswamy V, Shih DJH, Holgado BL, Farooq H, Donovan LK, Garzia L, Agnihotri S, Kiehna EN, Mercier E, Mayoh C, Papillon-Cavanagh S, Nikbakht H, Gayden T, Torchia J, Picard D, Merino DM, Vladoiu M, Luu B, Wu X, Daniels C, Horswell S, Thompson YY, Hovestadt V, Northcott PA, Jones DTW, Peacock J, Wang X, Mack SC, Reimand J, Albrecht S, Fontebasso AM, Thiessen N, Li Y, Schein JE, Lee D, Carlsen R, Mayo M, Tse K, Tam A, Dhalla N, Ally A, Chuah E, Cheng Y, Plettner P, Li HI, Corbett RD, Wong T, Long W, Loukides J, Buczkowicz P, Hawkins CE, Tabori U, Rood BR, Myseros JS, Packer RJ, Korshunov A, Lichter P, Kool M, Pfister SM, Schüller U, Dirks P, Huang A, Bouffet E, Rutka JT, Bader GD, Swanton C, Ma Y, Moore RA, Mungall AJ, Majewski J, Jones SJM, Das S, Malkin D, Jabado N, Marra MA, Taylor MD (2017) Spatial heterogeneity in medulloblastoma. Nat Genet 49: 780–788.
Nieto MA, Huang RY-J, Jackson RA, Thiery JP (2016) EMT: 2016. Cell 166: 21–45.
Procopio M-G, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo S-H, Goruppi S, Menietti E, Ostano P, Ala U, Provero P, Hoetzenecker W, Neel V, Kilarski WW, Swartz MA, Brisken C, Lefort K, Dotto GP (2015) Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 17: 1193–1204.
Reifenberger G, Weber RG, Riehmer V, Kaulich K, Willscher E, Wirth H, Gietzelt J, Hentschel B, Westphal M, Simon M, Schackert G, Schramm J, Matschke J, Sabel MC, Gramatzki D, Felsberg J, Hartmann C, Steinbach JP, Schlegel U, Wick W, Radlwimmer B, Pietsch T, Tonn JC, von Deimling A, Binder H, Weller M, Loeffler M, German Glioma Network (2014) Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int J Cancer 135: 1822–1831.
Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B (2008) Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs 17: 1533–1545.
Scheel C, Weinberg RA (2012) Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22: 396–403.
Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G, Zarif JC, Epstein T, Gatenby R, McCartney A, Elisseeff JH, Mooney SM, An SS, Pienta KJ (2014) Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6: 130–143.
Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G, Zarif JC, Epstein T, Gatenby R, McCartney A, Elisseeff JH, Mooney SM, An SS, Pienta KJ (2015) Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6: 130–143.
Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D, Nabilsi NH, Kladde MP, Suslov O, Brabletz S, Brabletz T, Reynolds BA, Steindler DA (2013) The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5: 1196–1212.
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65: 5–29.
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29: 4741–4751.
Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, Aranda S, Palomo S, McCormick F, Izpisúa-Belmonte JC, de la Pompa JL (2004) Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 18: 99–115.
Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN Cancer Genome Atlas Research Network (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110.
Wang J, Sullenger BA, Rich JN (2012) Notch signaling in cancer stem cells. Adv Exp Med Biol 727: 174–185.
Wenger RH, Stiehl DP, Camenisch G (2005) Integration of oxygen signaling at the consensus HRE. Sci STKE 2005: re12.
Xie Q, Wu Q, Kim L, Miller TE, Liau BB, Mack SC, Yang K, Factor DC, Fang X, Huang Z, Zhou W, Alazem K, Wang X, Bernstein BE, Bao S, Rich JN (2016) RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J Clin Invest 126: 2757–2772.
Yan W, Zhang W, You G, Bao Z, Wang Y, Liu Y, Kang C, You Y, Wang L, Jiang T (2012) Correlation of IDH1 mutation with clinicopathologic factors and prognosis in primary glioblastoma: a report of 118 patients from China. PLoS One 7: e30339.
Zhang T, Guo L, Creighton CJ, Lu Q, Gibbons DL, Yi ES, Deng B, Molina JR, Sun Z, Yang P, Yang Y (2016) A genetic cell context-dependent role for ZEB1 in lung cancer. Nat Commun 7: 12231.
Acknowledgements
UDK expresses his special thanks to HJS, Department of Neurosurgery, Dusseldorf, Germany for stimulating discussions and his continuous support. We thank MS Carro, Department of Neurosurgery, Freiburg, Germany for the uncomplicated exchange of cell models. JM and UDK are supported by the research commission of the medical faculty and the Strategic Research Fund (both from Heinrich-Heine University Dusseldorf). This work was supported by the Düsseldorf School of Oncology (funded by the Comprehensive Cancer Center Düsseldorf/Deutsche Krebshilfe and the Medical Faculty HHU Düsseldorf to KK and VM). MR is supported by the Deutsche Forschungsgemeinschaft (DFG; RE 2857/2-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Maciaczyk, D., Picard, D., Zhao, L. et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br J Cancer 117, 102–112 (2017). https://doi.org/10.1038/bjc.2017.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.157
Keywords
This article is cited by
-
The role of the ZEB1–neuroinflammation axis in CNS disorders
Journal of Neuroinflammation (2022)
-
Thermal stress involved in TRPV2 promotes tumorigenesis through the pathways of HSP70/27 and PI3K/Akt/mTOR in esophageal squamous cell carcinoma
British Journal of Cancer (2022)
-
Hypoxia in solid tumors: a key promoter of cancer stem cell (CSC) resistance
Journal of Cancer Research and Clinical Oncology (2020)
-
Targeting the Notch1 oncogene by miR-139-5p inhibits glioma metastasis and epithelial-mesenchymal transition (EMT)
BMC Neurology (2018)
-
Ars2 promotes cell proliferation and tumorigenicity in glioblastoma through regulating miR-6798-3p
Scientific Reports (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}