
Abstract
Background:
In B-cell precursor acute lymphoblastic leukaemia (B-ALL), the identification of additional genetic alterations associated with poor prognosis is still of importance. We determined the frequency and prognostic impact of somatic mutations in children and adult cases with B-ALL treated with Spanish PETHEMA and SEHOP protocols.
Methods:
Mutational status of hotspot regions of TP53, JAK2, PAX5, LEF1, CRLF2 and IL7R genes was determined by next-generation deep sequencing in 340 B-ALL patients (211 children and 129 adults). The associations between mutation status and clinicopathological features at the time of diagnosis, treatment outcome and survival were assessed. Univariate and multivariate survival analyses were performed to identify independent prognostic factors associated with overall survival (OS), event-free survival (EFS) and relapse rate (RR).
Results:
A mutation rate of 12.4% was identified. The frequency of adult mutations was higher (20.2% vs 7.6%, P=0.001). TP53 was the most frequently mutated gene (4.1%), followed by JAK2 (3.8%), CRLF2 (2.9%), PAX5 (2.4%), LEF1 (0.6%) and IL7R (0.3%). All mutations were observed in B-ALL without ETV6-RUNX1 (P=0.047) or BCR-ABL1 fusions (P<0.0001). In children, TP53mut was associated with lower OS (5-year OS: 50% vs 86%, P=0.002) and EFS rates (5-year EFS: 50% vs 78.3%, P=0.009) and higher RR (5-year RR: 33.3% vs 18.6% P=0.037), and was independently associated with higher RR (hazard ratio (HR)=4.5; P=0.04). In adults, TP53mut was associated with a lower OS (5-year OS: 0% vs 43.3%, P=0.019) and a higher RR (5-year RR: 100% vs 61.4%, P=0.029), whereas JAK2mut was associated with a lower EFS (5-year EFS: 0% vs 30.6%, P=0.035) and a higher RR (5-year RR: 100% vs 60.4%, P=0.002). TP53mut was an independent risk factor for shorter OS (HR=2.3; P=0.035) and, together with JAK2mut, also were independent markers of poor prognosis for RR (TP53mut: HR=5.9; P=0.027 and JAK2mut: HR=5.6; P=0.036).
Conclusions:
TP53mut and JAK2mut are potential biomarkers associated with poor prognosis in B-ALL patients.
Similar content being viewed by others


Main
B-cell precursor acute lymphoblastic leukaemia (B-ALL) is a malignancy of lymphoid progenitor cells, characterised by large biological and clinical heterogeneity (Roberts and Mullighan, 2015). ALL is the most frequent childhood cancer and also accounts for ∼25% of adult acute leukaemias (Bhojwani et al, 2015). High-risk B-ALL disease is more likely with adults than children, and the long-term disease-free adult survival rates are <40%, despite intensive chemotherapy and/or allogeneic stem cell transplantation treatments. This is in stark contrast to pediatric ALL, in which refined treatment regimens have resulted in cure rates approaching 80% (Paulsson et al, 2008; Bhojwani et al, 2015). However, in spite of this high cure rate, some children with ALL have a poor outcome, whereby 15% of them die from ALL relapses (Gowda and Dovat, 2013).
To date, the prognosis of B-ALL patients has focused mainly on clinical, haematological and genetic factors, such as age, leukocyte count at diagnosis, percentage of blast in peripheral blood, immunophenotype, central nervous system involvement, cytogenetic and molecular alterations and the presence of minimal residual disease (MRD), the latter two characteristics being most strongly associated with prognosis (Izraeli, 2010; Pui et al, 2011; Salari et al, 2014; Schrappe, 2014). However, ∼30% of pediatric and 50% of adult ALL patients lack defined genetic hallmarks of biological and clinical significance (Bungaro et al, 2009; Dawson et al, 2011; Forero-Castro et al, 2016a; Forero-Castro et al, 2016b). Somatic mutations are hallmarks of lymphoid malignancies, and each genetic subtype harbours hidden mutations that are strong independent predictors of outcome (Iacobucci et al, 2012).
In recent years, the use of next-generation sequencing (NGS) has greatly increased the ability to identify somatic mutations with clinical impact in both child and adult B-ALL. These mutations involved genes associated with RAS signalling (48%; e.g., NRAS, KRAS, PTPN11, FLT3, BRAF and NF1), B-cell differentiation and development (18%; e.g., PAX5, IKZF1, EBF1, VPREB1), JAK/STAT signalling (11%; e.g., JAK1, JAK2, IL7R and CRLF2), cell cycle regulation and tumour suppression (6%; e.g., TP53, RB1, CDKN2A/B, PTEN and BTG1), and non-canonical pathways (9%, e.g., ETV6, CREBBB and TBL1XR1) (Harrison, 2011; Mullighan, 2011; Pui et al, 2011; Roberts and Mullighan, 2011; Iacobucci et al, 2012; Loh and Mullighan, 2012; Gowda and Dovat, 2013; Inaba et al, 2013; Chiaretti et al, 2014a; Chiaretti et al, 2014b; Woo et al, 2014). However, the prognostic impact of these mutations as predictors of clinical course, outcome and response to therapy is still being explored.
Herein, the frequency and clinical relevance of somatic mutations within a selected custom panel of six genes, TP53, JAK2, IL7R, PAX5, LEF1 and CRLF2 exons, was examined. Targeted exonic regions with known mutational hotspots (Harrison, 2011; Iacobucci et al, 2012; Inaba et al, 2013; Roberts and Mullighan, 2015), were analysed by amplicon-based NGS in 340 B-ALL patients. We demonstrated that mutations in TP53 and JAK2 have a negative impact on the outcome of pediatric and adult patients. This supports their role as prognostic biomarkers, and suggests that if assessed at diagnosis, they might contribute to a better risk stratification of B-ALL patients.
Materials and methods
Patients and data collection
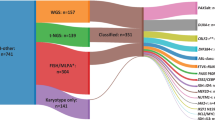
A total of 340 B-ALL patients referred from 22 Spanish centres to the Hematology Service at the Salamanca University Hospital, Spain, between February 1996 and February 2015 were eligible for this study. Two hundred and eleven of the patients were children (62.1%, <18 years) and 129 were adults (37.9%, ⩾18 years).
The diagnosis of B-ALL was based on morphological, immunophenotypic and genetic features of leukaemic blast cells, as described previously (Pui and Evans, 1998). Conventional cytogenetic analyses, fluorescent in situ hybridisation, demographic information, clinical characteristics, risk classification, frontline therapy protocol, response to therapy and survival were recorded. The criteria for defining and categorising the primary chromosomal abnormalities were established according to a previous study (Forero-Castro et al, 2016a), and are showed in Supplementary Material file 1. Patients were treated according to PETHEMA (Programa Español de Tratamientos en Hematología) and SEHOP (Sociedad Española de Hematología y Oncología Pediátrica) risk-adapted protocols. The study was approved by the local ethical committee, ‘The Comité Ético de Investigación Clínica del área de salud de Salamanca’, at the Hospital Universitario de Salamanca. Written informed consent was obtained from each patient or legal guardians before patients entered the study.
DNA isolation
Amplicon-based NGS was performed on all 340 samples obtained from untreated patients at diagnosis. Genomic DNA was extracted from frozen bone marrow or fixed peripheral blood cell samples with the QIAmp DNA Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions.
Next-generation amplicon deep-sequencing assay
An amplicon-based NGS assay was applied, using Titanium amplicon chemistry on a 454-GS Junior DNA Sequencing Platform (454 Life Sciences, Branford, CT, USA). With this approach, preconfigured custom 96-well primer plates containing lyophilised primer pairs (Roche, Branford, CT, USA) were used to prepare the amplicon library following the IRON-II Study procedures from the European Leukaemia Network group (Kohlmann et al, 2011). Nineteen hotspot exons were amplified: TP53 (E4-E11), JAK2 (E12-E16), PAX5 (E2-E3), LEF1 (E2-E3), CRLF2 (E6) and IL7R (E5). The size of amplicons ranged from 304 to 431 bp including the adaptor sequences (see Supplementary Material File 1: Supplementary Table S1 for primer sequences). Information about amplicon library pooling, purification, emulsion PCR, sequencing, validation of variants, data processing and analysis, is presented in the Supplementary Material File 1.
CRLF2 gene expression analysis
In a subset of 97 B-ALL patients (81 children and 16 adults) the expression levels of CRLF2 were evaluated by quantitative RT-PCR assays. CRLF2-overexpressed samples were identified as previously described (Yoda et al, 2010) (see Supplementary Material file 1 for further details).
Statistical methods
Continuous variables were summarised as their median and range; categorical variables were described as the frequency and percentage of subjects in each category. Associations between the presence of mutations and the values of clinical parameters were investigated. Continuous variables were analysed by the non-parametric Mann–Whitney U-test. The χ2 or Fisher’s exact tests were used to identify significant associations between dichotomous variables, as appropriate. All tests were two-sided and values of P<0.05 were considered to be significant. Kaplan–Meier analysis (log-rank test) was used to assess the relationship between mutations and overall survival (OS), event-free survival (EFS) and relapse rate (RR). Survival rates were given as probabilities of survival at 5 years, with a 95% confidence interval (CI). Pediatric and adult patients were analysed separately. Survival criteria are shown in the Supplementary Material File 1. Clinical and genetic variables were first analysed by univariate analysis, and those with a P-value up to 0.05 were included in multivariate analysis. A multivariate Cox proportional hazards regression model was used to estimate the hazard ratio (HR) and 95% CI of risk factors. Specifically simultaneous regression, which SPSS calls the Enter method was used. Analyses were performed using SPSS version 22.0 (IBM).
Results
Patient characteristics
Table 1 shows the clinical characteristics of the pediatric and adult patients with B-ALL included in this study. The median age was 12 years (range 0–84 years); childhood patients were aged from 0 to 17 years (median 5 years) while adult patients were aged from 18 to 84 years (median 45 years). The median percentage of blast counts in bone marrow was 90% (range, 35–100%) and 30.6% of patients showed normal cytogenetics. The presence of poor prognosis abnormalities t(9;22) (Ph+), t(v;11q23) and hypodiploidy, were more frequent in adult than in childhood B-ALL (48.8% vs 8.1%, P<0.0001). In addition, there was a higher prevalence of highly hyperdiploid cases (>50 chromosomes) in the pediatric compared with the adult group (15.6% vs 3.9%, P=0.001). The median follow-up of the whole series was 60 months (range, 2–186 months). The children had higher rates of 5-year OS (85.3% vs 40.5%, P<0.0001), EFS (77.7% vs 28.6%, P<0.0001) and relapse (RR) (16.1% vs 48.1%, P<0.0001) compared with adults.
Frequency and characterisation of molecular mutations in B-ALL
Thirty different mutations were observed in 42 out of the 340 patients at diagnosis (12.4%). TP53 was the most frequently mutated gene, being present in 4.1% of patients, followed by JAK2 (3.8%), CRLF2 (2.9%), PAX5 (2.4%), LEF1 (0.6%) and IL7R (0.3%). The mutations were more frequent in adults than in children (20.2% vs 7.6%, P=0.001). The detailed frequency of mutations in children and adults was TP53 (2.4% vs 7.0%, P=0.038), CRLF2 (0.5% vs 7.0%, P=0.001), JAK2 (2.4% vs 6.2%, P=0.086), PAX5 (1.4% vs 3.9%, P=0.162), LEF1 (0.5% vs 0.8%, P=1.0) and IL7R (0.5% vs 0%, P=1.0) (Supplementary Material File 1: Supplementary Figures S1 and S2).
The clinical characteristics, cytogenetic subgroups, somatic mutations with their respective mutational burden, risk classification, frontline therapy protocol used, response to therapy, survival and clinical status of each pediatric and adult patient are summarised in Supplementary Material File 1: Supplementary Table S2. Most mutations (88.1%) were detected in B-ALL cases lacking recurrent fusion genes. In fact, the presence of mutations in patients with normal cytogenetics was more frequently observed in both childhood (37.5%) and adult (46.2%) B-ALL cohort (P=0.002) (Supplementary Material File 1: Supplementary Table S3). It should be noted that only one pediatric patient (ID10) with TP53mut harboured the TCF3(E2A)-PBX1 fusion and four patients (one child, ID13 and three adults, ID25, ID26 and ID34) with PAX5mut, TP53mut and/or CRLF2mut carried KMT2A(MLL)-R.
Supplementary Material File 1: Supplementary Table S4 details the frequency of secondary somatic mutations in hotspot regions of TP53, JAK2, PAX5, LEF1, CRLF2 and IL7R genes classified by primary chromosomal abnormalities in children and adult patients with B-ALL. In the entire childhood cohort of B-ALL, all 16 mutations were exclusively detected in the subgroup of patients without ETV6-RUNX1 (TEL-AML1) translocation (P=0.047). Moreover, in the entire adult cohort of B-ALL, all 26 mutations were exclusively detected in the subgroup of patients without BCR-ABL1 translocation (P<0.0001). Thus, none of the cases with secondary somatic mutations showed ETV6-RUNX1 (TEL-AML1) or BCR-ABL1 translocations as primary chromosomal abnormalities. There were no other correlations between the primary chromosomal abnormality and the spectrum of mutations observed.
Supplementary Material File 1: Supplementary Table S5 details the primary chromosomal abnormalities evaluated by conventional and molecular cytogenetics, and describes the secondary somatic mutations according to the number of mutations per case, their mutational burden, gene-exon mutated and type of mutation observed in children and adult patients with B-ALL. The median mutational burden was 24.5% (range, 2–97%) (16.5% children and 28% adults, P=0.730). It is of particular note that 25.9% of mutations detected showed mutation loads of ⩽10% (23.5% children and 27% adults, P=1.0), so these variants would not have been detected by the Sanger capillary sequencing method (Supplementary Material File 1: Supplementary Table S2).
Twenty-two of the 30 different mutations found were previously described in the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) and/or the IARC TP53 database (http://p53.iarc.fr/p53Sequences.aspx) (Leroy et al, 2014) but we identified eight undescribed mutations in those genes (one in each gene under study and three novel missense mutations in PAX5). Supplementary Material File 1: Supplementary Table S6 showed the protein domain affected by these mutations. The sequence analysis revealed 24 missense mutations, three deletion-insertions, two splicing mutations and one frameshift mutation. As shown in Supplementary Material File 1: Supplementary Figures S1 and S2, TP53 mutations were generally distributed across several exons, with missense mutations being predominantly found in evolutionarily conserved regions of this gene. The other mutated genes showed recurrent mutations: CRLF2 (p.F232C, nine patients), JAK2 (p.R683G, nine patients and p.R683S, nine patients) and PAX5 (p.P80R, four patients).
NGS enabled identification of the co-occurrence of mutations and prediction of the presence or absence of distinct subclones harbouring different mutations
Eleven out of the 42 patients with mutations (26.2%) concomitantly harboured more than one mutation, which were more frequently observed in adult than in pediatric patients (38.5% vs 6.3%, P=0.030). Ten cases had two and one adult case had three mutations in the genes investigated. All of them were orthogoncally validated by other methods such as Sanger sequencing, re-sequencing run and/or the IonTorrent sequencer system. Supplementary Material File 1: Supplementary Table S7 shows the distribution of mutations identified in these 11 patients and Supplementary Material File 1: Supplementary Table S2 details their clinical characteristics, frontline therapy and outcome. In four cases (child: ID5 and adults: ID40, ID26 and ID27), the mutations occurred in the same gene and in the same amplicon (JAK2, PAX5 or TP53), while one adult patient (ID39) showed two different mutations within two amplicons of the same gene (TP53). In five adult cases, mutations occurred in different genes: three patients (ID17, ID21 and ID42) harboured concomitant JAK2 and CLFR2 mutations, while one patient (ID25) had TP53/PAX5, and other (ID19) CRFL2/LEF1 mutations. Finally, one adult patient (ID31) harboured three concomitant mutations in JAK2 and CRLF2, of which two mutations were detected in the same JAK2 amplicon.
The amplicon-NGS allowed separate subclones to be distinguished in two adult patients (ID31 and ID40, Supplementary Material File 1: Supplementary Figure S3 and Supplementary Table S2) with the same combination of JAK2 mutations located in exon 16 (JAK2-E16, p.R683G and p.R683S). In these cases the mutated codon 683 was not found concomitantly within the same sequencing read, but was separated across distinct individual reads. In accordance with the differential mutational loads between the two JAK2 mutations, the sequencing analysis demonstrated that the dominant clone harboured the p.R683G mutation (c.2047A>G), whereas the minority clone harboured the p.R683S mutation (c.2049A>T). In both cases, the c.2047A>G mutation was always present at a higher percentage with respect to the c.2049A>T mutation (patient ID31: 26% and 2%; and patient ID40: 38% and 4%) and the minority subclone was present with a mutational burden of <5% of leukaemic cells. Finally, one of these patients (patient ID31) harboured a third low-percentage mutation in the CRLF2 gene (p.F232C in 10% of leukaemic cells).
In addition, amplicon-NGS made it possible to identify clonal heterogeneity in three patients (one child and two adults) who carried a double mutation within the same amplicon. In the first patient (ID27-adult) who harboured two distinct mutations in the same amplicon (PAX5-E03, c.215A>G and c.239C>G), 454 deep-sequencing allowed the presence of four lines to be discriminated; one minority and germline (PAX5wt: 8.8%), one dominant clone with c.215A>G mutation (51.4%), one subclone with c. 239C>G mutation (36.6%) and one minority subclone (3.2%) with a double mutation in the PAX5-E03 gene (c.215A>G and c. 239C>G) (Supplementary Material File 1: Supplementary Figure S4). In the second patient (ID5-child) who carried two mutations in the JAK2-E16 gene, three lines were identified: one germline (JAK2wt: 84%), one subclone with c.2044_2045insGGACCTCCTCCCTCC mutation (12%) and one minority subclone with c.2049A>T mutation (4%) (Supplementary Material File 1: Supplementary Figure S5). Finally, three independent lines were identified in the third patient (ID26-adult) presenting two mutations in the TP53-E08 gene: one germline (TP53wt:83.4%) line, and two minority lines, one subclone with c.841G>A mutation (10%) and one subclone with c.845G>C mutation (6.6%) (Supplementary Material File 1: Supplementary Figure S6).
Mutated genes associated with clinical and prognostic features
Next, mutations were associated with cytogenetic subtypes, clinical features and risk factors commonly used to stratify pediatric and adult B-ALL patients (Supplementary Material File 1: Supplementary Table S8 and Supplementary Table S9). Thus, the presence of mutations in any of the genes analysed was associated with children stratified in the high-risk group (P=0.018). Mutations in the TP53 gene were associated with poor response to frontline therapy due to refractoriness or relapse events (P=0.032) (Supplementary Material File 1: Supplementary Table S8). Meanwhile, in the adult cohort, all 26 mutated cases were exclusively detected in BCR-ABL1-negative cases (P<0.0001). The presence of JAK2mut was associated with poor-prognosis frontline therapy (P=0.031) (Supplementary Material File 1: Supplementary Table S9).
Gene mutations are related to a worse outcome
In the survival analysis of the children, significantly lower OS (5-year OS: 50% vs 86%, P=0.002) and EFS rates (5-year EFS: 50% vs 78.3%, P=0.009) and higher RR (5-year RR: 33.3% vs 18.6% P=0.037), were observed in patients with TP53mut compared with patients without TP53mut (Figure 1 and Supplementary Material file 1: Supplementary Table S10). The present study also confirmed in pediatric cohort the well-known associations of particular clinical and biological variables with worse prognosis, such as the high-risk group (OS: P<0.0001, EFS: P<0.0001 and RR: P=0.002), MRD ⩾0.01% (OS: P=0.004 and EFS: P=0.027), WBC ⩾30 × 109 l−1 (OS: P=0.004), poor-risk cytogenetics due to the presence of t(9;22), t(v;11q23) or a hypodiploid karyotype (OS: P<0.0001, EFS: P<0.0001 and RR: P=0.016), age over 10 years (OS: P<0.0001, EFS: P<0.0001 and RR: P<0.0001), and a Pro-B phenotype (OS: P<0.0001 and EFS: P=0.004) (Supplementary Material file 1: Supplementary Table S10). Multivariate analysis of the group of children showed that TP53mut was an independent risk factor associated with significantly higher RR (HR=4.5; 95% CI 1.1–19.2, P=0.04) (Table 2).

Kaplan–Meier curves for OS, EFS and RR of the whole cohort of children with B-ALL. (A–C) OS, EFS and RR in patients bearing TP53mut.
In the group of adults, TP53mut had a negative effect on OS (5-year OS: 0% vs 43.3%, P=0.019) and was associated with a higher RR (5-year RR: 100.0% vs 61.4%, P=0.029), and JAK2mut was related with a lower EFS (5-year EFS: 0% vs 30.6%, P=0.035) and a higher RR (5-year RR: 100% vs 60.4%, P=0.002) (Figure 2 and Supplementary Material File 1: Supplementary Table S11). The NGS analysis also confirmed the clinical parameters commonly associated with shorter OS, EFS and RR in adults, such as MRD ⩾0.01% (RR: P=0.003 and EFS: P=0.023), WBC ⩾30 × 109 l−1 (RR: P=0.024 and EFS: P=0.008), poor-risk cytogenetic abnormalities (OS: P=0.013 and EFS: P=0.025) and age ⩾55 years (OS: P=0.001, RR: P=0.027 and EFS: P=0.002). Supplementary Material File 1: Supplementary Table S11 shows these clinical parameters commonly associated with shorter survival in adults. Multivariate analysis of the whole cohort of ALL adults indicated that the presence of TP53mut was an independent risk factor associated with significantly shorter OS (HR=2.3; 95% CI 1.1–5.1, P=0.035). Moreover, TP53mut (HR=5.9; 95% CI 1.2–28.6, P=0.027) and JAK2mut (HR=5.6; 95% CI 1.1–28.1, P=0.036) retained their independent prognostic significance in multivariate analysis regarding for RR (Table 3).

Kaplan–Meier curves for OS, EFS and RR of the whole cohort of adults with B-ALL. (A and B) OS and RR in patients bearing TP53mut. (C and D) EFS and RR in patients with JAK2mut.
Finally, it is worth mentioning that there were no associations between the mutational burden and the cytogenetic, clinical and prognostic parameters of the pediatric and adult cohorts with B-ALL. In the same way, the survival rates of patients who carried small clones with ⩽10% of mutational burden did not have significant differences with respect to those who carried clone sizes >10% (children: 5-year OS: 50% vs 72.7%, P=0.544; 5-year EFS: 50% vs 72.7%, P=0.606 and 5-year RR: 50.0% vs 20%, P=0.426; adults: 5-year OS: 0% vs 31.3%, P=0.453 and 5-year EFS: 0% vs 12.5%, P=0.710). Particularly, for TP53 and JAK2 mutations, there was no difference of mutational burden on outcome and survival parameters.
CRLF2 overexpression is associated with JAK mutations
Overexpression of the CRLF2 gene (CRLF2oe) was observed in 17.5% of B-ALL patients by quantitative RT-PCR assays. There were no significant differences in the CRLF2oe between child and adult B-ALL patients (17.3% vs 18.8%, P=1.0). CRLF2oe was common only in B-ALL cases that lack rearrangements of TEL-AML1, KMT2A(MLL), TCF3(E2A)-PBX1, and BCR-ABL (P=0.007). Supplementary Material File 1: Supplementary Table S2 shows the results obtained from the analysis of CRLF2 expression in each of the patients with mutations. The presence of JAK2 mutations was associated with CRLF2oe (P=0.023). Furthermore, neither the clinical nor prognostic features were associated with CRLF2oe in both childhood and adult B-ALL patients. However, the survival analysis of whole cohort of adults with B-ALL showed that the presence of CRLF2oe had a negative effect on OS (5-year OS: 0% vs 53.8%, P=0.005) and EFS rates (5-year EFS: 0% vs 33.3%, P=0.006).
Discussion
The presence of gene mutations is a hallmark of B-ALL. In this study, we evaluated 340 B-ALL cases at diagnosis (211 children and 129 adults) to address the incidence and prognostic impact of TP53, JAK2, IL7R, PAX5, LEF1 and CRLF2 mutations by NGS. The incidence (12.4%) and frequency of mutations were higher in adults than in children according with the better prognosis of pediatric B-ALL. Most mutations (88.1%) were detected in B-ALL cases lacking recurrent fusion genes. The incidence and frequency of mutations detected in our study were similar to observations made in previous ALL studies (Mullighan, 2011; Pui et al, 2011; Roberts and Mullighan, 2011; Iacobucci et al, 2012; Loh and Mullighan, 2012; Gowda and Dovat, 2013; Inaba et al, 2013; Chiaretti et al, 2014a, 2014b; Woo et al, 2014). The use of the NGS strategy further allowed the identification of clonal heterogeneity in B-ALL patients. A negative impact of the presence of TP53 and JAK2 mutations on the OS, RR and EFS in patients with adult and childhood B-ALL was found in the present study. Therefore, deep sequencing may ultimately be a better guide to treatment decisions for B-ALL patients.
Among 30 mutations identified at diagnosis, 22 have previously been described in B-ALL databases (Bercovich et al, 2008; Mullighan et al, 2009b; Roll and Reuther, 2010; Yoda et al, 2010; Mullighan et al, 2011; Izraeli et al, 2014; Leroy et al, 2014), confirming these mutated exons to be hotspot regions in leukaemia. Recurrent mutations in CRLF2 (p.F232C), JAK2 (p.R683G and p.R683S) and PAX5 (p.P80R) have also been observed in other cohorts of patients with ALL (Bercovich et al, 2008; Mullighan et al, 2009b; Roll and Reuther, 2010; Yoda et al, 2010; Mullighan et al, 2011; Izraeli et al, 2014). It is of particular note that we have found five undescribed mutations in those genes, three of which are missense mutations in PAX5. Mutations in the paired box (PAX) domain are predicted as deleterious, probably impairing the DNA-binding capability of this lymphoid transcription factor which is essential for normal B-cell development (Roberts et al, 2001). Also, two undescribed mutations were found in either IL7R or LEF1, the least frequently mutated genes in our series. Somatic gain-of-function mutations in IL-7R have been shown to act as oncogenes in T- and B-ALL (Shochat et al, 2011; Mazzucchelli et al, 2012). (Shochat et al, 2011). These IL-7R mutations are usually insertions of bases encoding a cysteine and a proline into the 6th exon of the extracellular domain, at the border with the transmembrane region, immediately before or after residue 244 (Mazzucchelli et al, 2012). It is well known that cysteines and prolines are essential for the constitutive activation of the receptor causing cytokine independent growth of mouse pro-B cells (Shochat et al, 2011).
In the case of TP53mut, 11 out of 13 patients were negative for fusion genes, consistent with the findings of a previous ALL cohort at diagnosis (Chiaretti et al, 2013). Only one pediatric patient (ID10) with the TCF3(E2A)-PBX1 fusion and one adult (ID26) with KMT2A(MLL)-R harboured TP53mut. It should be noted that a significant association between TP53mut and MLL/AFF1 translocations has been reported (Hof et al, 2011). Furthermore, two patients with low hypodiploid (ID9 and ID38) and two patients with near triploid karyotypes (ID22 and ID24) carried TP53 mutations. The high incidence of TP53 alterations (mutation, deletion) was previously associated with low hypodiploid/near triploid ALL, making it possible to distinguish this rare subset from other ALL subgroups including near haploid ALL. Furthermore, this high incidence can guide the prognosis of the disease because these alterations are usually associated with worse prognosis (Muhlbacher et al, 2014; Stengel et al, 2014). Therefore, the use of NGS could enable the prognostic B-ALL subgroups of patients to be refined.
Amplicon-based NGS was also able to detect variants with a low mutational burden, which have been identified in genomic databases (Bercovich et al, 2008; Mullighan et al, 2009b; Roll and Reuther, 2010; Yoda et al, 2010; Mullighan et al, 2011; Izraeli et al, 2014; Leroy et al, 2014) or found in previous studies in ALL patients (Shochat et al, 2011). Thus, our study confirmed that NGS is a suitable method for accurately detecting and quantifying a variety of mutations in important genes associated with pathogenesis and prognosis of B-ALL. In fact, 26.4% of variations described in our study had mutation loads of ⩽10%, underlining the high sensitivity of amplicon-based NGS for detecting small leukaemic subclones, commonly undetectable by conventional Sanger capillary sequencing. It is well known that mutational screening by bidirectional Sanger sequencing does not reveal the presence of mutant subclones representing less than 10–20% of leukaemic cells (Kastner et al, 2014).
One of the most important tasks in understanding clonal progression in leukaemia is to assess the nature and number of different subclones within an individual cancer (Mullighan et al, 2008; Grossmann et al, 2011; Landau et al, 2014) In agreement with the findings of previous studies (Kohlmann et al, 2010; Grossmann et al, 2011; Kohlmann et al, 2011; Kastner et al, 2014; Landau et al, 2014), we confirmed the complex clonal architecture of ALL. NGS technology allowed the detection of clonal heterogeneity in some of the analysed patients. This was achieved by identifying distinct subpopulations with a dominant leukaemic clone and their relative proportions in the total B-ALL cell population. The case ID27 (Supplementary Material File 1: Supplementary Figure S4) shows the presence of leukaemia subclones that were derived from a common tumour-initiating cell, whereas cases ID5 and ID26 (Supplementary Material File 1: Supplementary Figures S5 and S6) have clones with divergent mutations (Jan and Majeti, 2013). Further investigation with samples from relapsed ALL is needed to elucidate the effect of these clones and subclones on disease progression and whether they can provide clues to the cause of treatment failure (Meyer et al, 2013; Tzoneva et al, 2013; Lindqvist et al, 2015). Unfortunately these samples were not available in this study.
The present study showed that TP53 mutations are the mutations most frequently observed in B-ALL patients. The tumour suppressor gene TP53 has a crucial role in cell cycle regulation and apoptosis after DNA damage, and its role in tumorigenesis is well recognised in solid and haematological malignancies (Chiaretti et al, 2013). In our study, TP53mut was associated with poor outcome in the whole cohort of patients and when considering adults and children separately. These results are in agreement with those of previous studies in which TP53mut was associated with resistance to treatment and worse prognosis in several tumours (Olivier et al, 2010; Salmoiraghi et al, 2016). In addition, alterations of the TP53 gene were described as being important at relapse in childhood and adult ALL, in which they independently predict a high risk of treatment failure in a significant number of patients (Hof et al, 2011). The presence of TP53 mutations was also associated with a reduced response rate to induction therapy (Chiaretti et al, 2013) and a shorter survival (from time of diagnosis and from time of relapse), even after successful reinduction therapy (Diccianni et al, 1994). These lines of evidence, together with our results, highlight the importance of sequencing TP53 at diagnosis.
JAK2mut was also frequent in B-ALL. JAK2 is an important mediator of cytokine receptor signalling and has key roles in the hematopoietic and immune response. Constitutive signalling through JAK2 contributes to the proliferation of many cancers, including B-cell leukaemia and lymphomas (Ihle and Gilliland, 2007; Vainchenker and Constantinescu, 2013). The acquired JAK2mut (p.R683S and p.R683G) is presumed to be a biomarker for B-ALL (Bercovich et al, 2008; Mullighan, 2008; Mullighan et al, 2009b). Recent studies suggested that the amino acid residue p.R683 located in the linker between the N and C lobes of JH2 domain is important for maintaining the activity, structural stability and folding of JAK2 (Li et al, 2013). The mutations in p.R683 disrupted the structure of JH2 domain leading to JAK2 constitutive activation and induced growth factor-independent cell proliferation of the mouse Ba/F3 hematopoietic cell line (Mullighan, 2008; Li et al, 2013). In our study, two B-ALL patients with two independent clones each, displayed different mutations in the p.R683 residue (Bercovich et al, 2008). Even though the oncogenic effect of p.R683S and p.R683G mutations could be similar or identical, further research will be necessary to fully understand the biological relevance of this clonal heterogeneity. The development of JAK2 inhibitors that abrogates JAK/STAT activation may be a useful approach for treating patients harbouring these mutations (Mullighan et al, 2009b; Roberts and Mullighan, 2011).
We found that CRLF2oe is associated with JAK2 mutations in B-ALL in cases lacking recurrent gene fusions, according to previous reports (Mullighan et al, 2009a; Yoda et al, 2010; Harvey et al, 2010). In our study, the frequency of CRLF2oe found in children and adults was around 17%. While in children it was similar to that observed by Chen et al (2012), in adults was lower (Chiaretti et al, 2016) even though maintained its association with poor outcome in this group of patients. Therefore, CRLF2 quantification could be an important prognostic marker in adult B-ALL. Evidence from in vitro studies suggests that these proteins could cooperate to transform B-ALL cells as CRLF2 acts as a scaffold for JAK2mut signalling (Yoda et al, 2010), although the precise molecular mechanism remains undescribed. However, cells dependent on CRLF2 signalling are also sensitive to JAK2 inhibitors. Thus, ALL patients with CRLF2oe may benefit from future kinase inhibitor approaches (Roll and Reuther, 2010; Yoda et al, 2010). Future studies should be conducted to further elucidate JAK2/CRLF2 association and prognosis in B-ALL. In this sense, further assessment of gene rearrangements involving CRLF2 as IGH@-CRLF2 and P2RY8-CRLF2 should be performed to establish the prognostic significance of CRLF2oe and CRLF2 rearrangements across risk subgroups of B-ALL.
Conclusions
The amplicon NGS results indicate that the incidence of mutation in TP53, JAK2, IL7R, PAX5, LEF1 and CRLF2 is higher in adults than in children. All mutations were frequent in B-ALL cases without recurrent fusion genes. Given the negative influence on outcome, we suggest that TP53 and JAK2 status should be investigated at diagnosis, particularly in patients negative for recurrent fusion genes, for whom genetic-based prognostic stratification is still limited. Deep sequencing may ultimately guide treatment decisions better for B-ALL patients bearing TP53 and JAK2 mutations and could give rise to alternative therapeutic regimens.
Change history
11 July 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, Strehl S, Haas OA, Mann G, Binder V, Borkhardt A, Kempski H, Trka J, Bielorei B, Avigad S, Stark B, Smith O, Dastugue N, Bourquin JP, Tal NB, Green AR, Izraeli S (2008) Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484–1492.
Bhojwani D, Yang JJ, Pui CH (2015) Biology of childhood acute lymphoblastic leukaemia. Pediatr Clin North Am 62 (1): 47–60.
Bungaro S, Dell'Orto MC, Zangrando A, Basso D, Gorletta T, Lo Nigro L, Leszl A, Young BD, Basso G, Bicciato S, Biondi A, te Kronnie G, Cazzaniga G (2009) Integration of genomic and gene expression data of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks. Genes Chromosomes Cancer 48 (1): 22–38.
Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, Reshmi S, Cottrell CE, Reaman GH, Bowman WP, Carroll WL, Loh ML, Winick NJ, Hunger SP, Willman CL (2012) Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512–3522.
Chiaretti S, Brugnoletti F, Messina M, Paoloni F, Fedullo AL, Piciocchi A, Elia L, Vitale A, Mauro E, Ferrara F, De Fabritiis P, Luppi M, Ronco F, De Propris MS, Raponi S, Kronnie GT, Vignetti M, Guarini A, Foa R (2016) CRLF2 overexpression identifies an unfavourable subgroup of adult B-cell precursor acute lymphoblastic leukemia lacking recurrent genetic abnormalities. Leuk Res 41: 36–42.
Chiaretti S, Brugnoletti F, Tavolaro S, Bonina S, Paoloni F, Marinelli M, Patten N, Bonifacio M, Kropp MG, Sica S, Guarini A, Foa R (2013) TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica 98 (5): e59–e61.
Chiaretti S, Gianfelici V, Ceglie G, Foa R (2014a) Genomic characterization of acute leukemias. Med Princ Pract 23 (6): 487–506.
Chiaretti S, Zini G, Bassan R (2014b) Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr J Hematol Infect Dis 6 (1): e2014073.
Dawson AJ, Yanofsky R, Vallente R, Bal S, Schroedter I, Liang L, Mai S (2011) Array comparative genomic hybridization and cytogenetic analysis in pediatric acute leukemias. Curr Oncol 18 (5): e210–e217.
Diccianni MB, Yu J, Hsiao M, Mukherjee S, Shao LE, Yu AL (1994) Clinical significance of p53 mutations in relapsed T-cell acute lymphoblastic leukemia. Blood 84 (9): 3105–3112.
Forero-Castro M, Robledo C, Benito R, Abaigar M, Africa Martin A, Arefi M, Fuster JL, de Las Heras N, Rodriguez JN, Quintero J, Riesco S, Hermosin L, de la Fuente I, Recio I, Ribera J, Labrador J, Alonso JM, Olivier C, Sierra M, Megido M, Corchete-Sanchez LA, Ciudad Pizarro J, Garcia JL, Ribera JM, Hernandez-Rivas JM (2016a) Genome-wide DNA copy number analysis of acute lymphoblastic leukemia identifies new genetic markers associated with clinical outcome. PLoS One 11 (2): e0148972.
Forero-Castro M, Robledo C, Lumbreras E, Benito R, Hernandez-Sanchez JM, Hernandez-Sanchez M, Garcia JL, Corchete-Sanchez LA, Tormo M, Barba P, Menarguez J, Ribera J, Grande C, Escoda L, Olivier C, Carrillo E, Garcia de Coca A, Ribera JM, Hernandez-Rivas JM (2016b) The presence of genomic imbalances is associated with poor outcome in patients with burkitt lymphoma treated with dose-intensive chemotherapy including rituximab. Br J Haematol 172 (3): 428–438.
Gowda C, Dovat S (2013) Genetic targets in pediatric acute lymphoblastic leukemia. Adv Exp Med Biol 779: 327–340.
Grossmann V, Schnittger S, Schindela S, Klein HU, Eder C, Dugas M, Kern W, Haferlach T, Haferlach C, Kohlmann A (2011) Strategy for robust detection of insertions, deletions, and point mutations in CEBPA, a GC-rich content gene, using 454 next-generation deep-sequencing technology. J Mol Diagn 13 (2): 129–136.
Harrison CJ (2011) Key pathways as therapeutic targets. Blood 118 (11): 2935–2936.
Harvey RC, Mullighan CG, Chen IM, Wharton W, Mikhail FM, Carroll AJ, Kang H, Liu W, Dobbin KK, Smith MA, Carroll WL, Devidas M, Bowman WP, Camitta BM, Reaman GH, Hunger SP, Downing JR, Willman CL (2010) Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312–5321.
Hof J, Krentz S, van Schewick C, Korner G, Shalapour S, Rhein P, Karawajew L, Ludwig WD, Seeger K, Henze G, von Stackelberg A, Hagemeier C, Eckert C, Kirschner-Schwabe R (2011) Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185–3193.
Iacobucci I, Papayannidis C, Lonetti A, Ferrari A, Baccarani M, Martinelli G (2012) Cytogenetic and molecular predictors of outcome in acute lymphocytic leukemia: recent developments. Curr Hematol Malig Rep 7 (2): 133–143.
Ihle JN, Gilliland DG (2007) Jak2: normal function and role in hematopoietic disorders. Curr Opin Genet Dev 17 (1): 8–14.
Inaba H, Greaves M, Mullighan CG (2013) Acute lymphoblastic leukaemia. Lancet 381 (9881): 1943–1955.
Izraeli S (2010) Application of genomics for risk stratification of childhood acute lymphoblastic leukaemia: from bench to bedside? Br J Haematol 151 (2): 119–131.
Izraeli S, Shochat C, Tal N, Geron I (2014) Towards precision medicine in childhood leukemia—insights from mutationally activated cytokine receptor pathways in acute lymphoblastic leukemia. Cancer Lett 352 (1): 15–20.
Jan M, Majeti R (2013) Clonal evolution of acute leukemia genomes. Oncogene 32 (2): 135–140.
Kastner R, Zopf A, Preuner S, Proll J, Niklas N, Foskett P, Valent P, Lion T, Gabriel C (2014) Rapid identification of compound mutations in patients with Philadelphia-positive leukaemias by long-range next generation sequencing. Eur J Cancer 50 (4): 793–800.
Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, Dicker F, Schnittger S, Dugas M, Kern W, Haferlach C, Haferlach T (2010) Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 28 (24): 3858–3865.
Kohlmann A, Klein HU, Weissmann S, Bresolin S, Chaplin T, Cuppens H, Haschke-Becher E, Garicochea B, Grossmann V, Hanczaruk B, Hebestreit K, Gabriel C, Iacobucci I, Jansen JH, te Kronnie G, van de Locht L, Martinelli G, McGowan K, Schweiger MR, Timmermann B, Vandenberghe P, Young BD, Dugas M, Haferlach T (2011) The Interlaboratory RObustness of Next-generation sequencing (IRON) study: a deep sequencing investigation of TET2, CBL and KRAS mutations by an international consortium involving 10 laboratories. Leukemia 25 (12): 1840–1848.
Landau DA, Carter SL, Getz G, Wu CJ (2014) Clonal evolution in hematological malignancies and therapeutic implications. Leukemia 28 (1): 34–43.
Leroy B, Anderson M, Soussi T (2014) TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat 35 (6): 672–688.
Li F, Guo HY, Wang M, Geng HL, Bian MR, Cao J, Chen C, Zeng LY, Wang XY, Wu QY (2013) The effects of R683S (G) genetic mutations on the JAK2 activity, structure and stability. Int J Biol Macromol 60: 186–195.
Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, Overnas E, Dahlberg J, Wahlberg P, Henriksson N, Abrahamsson J, Frost BM, Grander D, Heyman M, Larsson R, Palle J, Soderhall S, Forestier E, Lonnerholm G, Syvanen AC, Berglund EC (2015) The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum Mutat 36 (1): 118–128.
Loh ML, Mullighan CG (2012) Advances in the genetics of high-risk childhood B-progenitor acute lymphoblastic leukemia and juvenile myelomonocytic leukemia: implications for therapy. Clin Cancer Res 18 (10): 2754–2767.
Mazzucchelli RI, Riva A, Durum SK (2012) The human IL-7 receptor gene: deletions, polymorphisms and mutations. Semin Immunol 24 (3): 225–230.
Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, Tang Z, Zumbo P, Li S, Zavadil J, Levine RL, Cardozo T, Hunger SP, Raetz EA, Evans WE, Morrison DJ, Mason CE, Carroll WL (2013) Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290–294.
Muhlbacher V, Zenger M, Schnittger S, Weissmann S, Kunze F, Kohlmann A, Bellos F, Kern W, Haferlach T, Haferlach C (2014) Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93%. Genes Chromosomes Cancer 53 (6): 524–536.
Mullighan C, Petersdorf E, Davies SM, DiPersio J (2011) From trees to the forest: genes to genomics. Biol Blood Marrow Transplant 17 (1 Suppl): S52–S57.
Mullighan CG (2008) JAK2—a new player in acute lymphoblastic leukaemia. Lancet 372 (9648): 1448–1450.
Mullighan CG (2011) Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best Pract Res Clin Haematol 24 (4): 489–503.
Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, Mikhail FM, Meyer J, Carroll AJ, Williams RT, Cheng J, Heerema NA, Basso G, Pession A, Pui CH, Raimondi SC, Hunger SP, Downing JR, Carroll WL, Rabin KR (2009a) Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243–1246.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR (2008) Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322 (5906): 1377–1380.
Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, Devidas M, Atlas SR, Chen IM, Clifford RJ, Gerhard DS, Carroll WL, Reaman GH, Smith M, Downing JR, Hunger SP, Willman CL (2009b) JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA 106 (23): 9414–9418.
Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2 (1): a001008.
Paulsson K, Cazier JB, Macdougall F, Stevens J, Stasevich I, Vrcelj N, Chaplin T, Lillington DM, Lister TA, Young BD (2008) Microdeletions are a general feature of adult and adolescent acute lymphoblastic leukemia: unexpected similarities with pediatric disease. Proc Natl Acad Sci USA 105 (18): 6708–6713.
Pui CH, Carroll WL, Meshinchi S, Arceci RJ (2011) Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29 (5): 551–565.
Pui CH, Evans WE (1998) Acute lymphoblastic leukemia. N Engl J Med 339 (9): 605–615.
Roberts EC, Deed RW, Inoue T, Norton JD, Sharrocks AD (2001) Id helix-loop-helix proteins antagonize pax transcription factor activity by inhibiting DNA binding. Mol Cell Biol 21 (2): 524–533.
Roberts KG, Mullighan CG (2011) How new advances in genetic analysis are influencing the understanding and treatment of childhood acute leukemia. Curr Opin Pediatr 23 (1): 34–40.
Roberts KG, Mullighan CG (2015) Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol 12 (6): 344–357.
Roll JD, Reuther GW (2010) CRLF2 and JAK2 in B-progenitor acute lymphoblastic leukemia: a novel association in oncogenesis. Cancer Res 70 (19): 7347–7352.
Salari F, Shahjahani M, Shahrabi S, Saki N (2014) Minimal residual disease in acute lymphoblastic leukemia: optimal methods and clinical relevance, pitfalls and recent approaches. Med Oncol 31 (11): 266.
Salmoiraghi S, Montalvo ML, Ubiali G, Tosi M, Peruta B, Zanghi P, Oldani E, Boschini C, Kohlmann A, Bungaro S, Intermesoli T, Terruzzi E, Angelucci E, Cavattoni I, Ciceri F, Bassan R, Rambaldi A, Spinelli O (2016) Mutations of TP53 gene in adult acute lymphoblastic leukemia at diagnosis do not affect the achievement of hematologic response but correlate with early relapse and very poor survival. Haematologica 101 (6): e245–e248.
Schrappe M (2014) Detection and management of minimal residual disease in acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2014 (1): 244–249.
Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, Cario G, Cazzaniga G, Kulozik AE, Stanulla M, Schrappe M, Biondi A, Basso G, Bercovich D, Muckenthaler MU, Izraeli S (2011) Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med 208 (5): 901–908.
Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A, Haferlach T, Haferlach C (2014) TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood 124 (2): 251–258.
Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe JM, Tallman MS, Paganin M, Basso G, Hof J, Kirschner-Schwabe R, Palomero T, Rabadan R, Ferrando A (2013) Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 19 (3): 368–371.
Vainchenker W, Constantinescu SN (2013) JAK/STAT signaling in hematological malignancies. Oncogene 32 (21): 2601–2613.
Woo JS, Alberti MO, Tirado CA (2014) Childhood B-acute lymphoblastic leukemia: a genetic update. Exp Hematol Oncol 3: 16.
Yoda A, Yoda Y, Chiaretti S, Bar-Natan M, Mani K, Rodig SJ, West N, Xiao Y, Brown JR, Mitsiades C, Sattler M, Kutok JL, DeAngelo DJ, Wadleigh M, Piciocchi A, Dal Cin P, Bradner JE, Griffin JD, Anderson KC, Stone RM, Ritz J, Foa R, Aster JC, Frank DA, Weinstock DM (2010) Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA 107 (1): 252–257.
Acknowledgements
We thank the physicians from the Spanish PETHEMA Group who contributed samples and clinical data, and the technical staff (Irene Rodríguez, Sara González, Luis Antonio Corchete, Teresa Prieto, María Ángeles Ramos, Almudena Martín, Ana Díaz, Ana Simón, María del Pozo, Isabel M Isidro, Vanesa Gutiérrez, Sandra Santos and Sandra Pujante) from the Cancer Research Center, Salamanca, for their assistance and advice. We also thank Dr Santiago Barrio (12 Octubre Hospital) for their initial work in NGS, C Blas (Jimenez Díaz Foundation) and Dr ML Toribio (CBM, CSIC) for providing clinical samples. This work was supported in part by a grant from the European Union’s Seventh Framework Programme (FP7/2007–2013) under Grant Agreement 306242-NGS-PTL, the Consejería de Educación, Junta de Castilla y León (HUS272U13, SA085U16 to JMHR and JCYL-EDU/346/2013 PhD scholarship to MHS), Fundación Castellano Leonesa de Hematología y Hemoterapia (FUCALHH 2013) to JMHR, Proyectos de Investigación del SACYL, Spain (BIO/SA31/13 and BIO/SA10/14) to RB; The Instituto de Salud Carlos III from Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund (ISCIII-FEDER) “Una manera de hacer Europa” Spanish Cancer Network (RD12/0036/0069 and RD12/0036/0061 to JM) and FIS grants PI15/01471 to JMHR and PI15/00032 to EFR. Universidad Pedagógica y Tecnológica de Colombia—Vicerrectoría de Investigación y Extensión (Grupo de Investigación en Ciencias Biomédicas UPTC—GICBUPTC, Escuela de Ciencias Biológicas) to MFC. Mutation analyses were performed using custom oligonucleotide primer plates designed as part of the IRON-II collaborative network of haematological laboratories applying 454 Life Sciencies amplicon-based NGS.
Declarations
Ethics approval and consent to participate: The study was approved by the local ethical committee, the Comité Ético de Investigación Clínica del área de salud de Salamanca, at the Hospital Universitario de Salamanca. Written informed consent was obtained from each patient or legal guardians before patients entered the study. Availability of data and material: The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Author contributions
MFC wrote the paper and edited the manuscript. MFC, IBM, SR and EFR recorded and analysed the clinical and biological data. MFC, CR, RB, MA, MHS, JMHS, MQA, IBM, MSV and IR designed, performed and analysed the NGS experiments. IBM, MSV and FAS performed the CRLF2 expression studies. JLF, MArf, NH, JNR, LH, JR, IR, JMSP, MC, MR, CD, EB, JM, JMR and JMHR provided patient samples and the clinical data. AK provided reagents and supported NGS analysis; all authors participated in discussions and critically reviewed the manuscript; JM, EFR, AK and JMHR analysed and interpreted the data, led and supervised the study and corrected and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Forero-Castro, M., Robledo, C., Benito, R. et al. Mutations in TP53 and JAK2 are independent prognostic biomarkers in B-cell precursor acute lymphoblastic leukaemia. Br J Cancer 117, 256–265 (2017). https://doi.org/10.1038/bjc.2017.152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.152
Keywords
This article is cited by
-
Mutations of epigenetic modifier genes predict poor outcome in adult acute lymphoblastic leukemia
Annals of Hematology (2024)
-
Consolidative allogeneic hematopoietic stem cell transplantation after chimeric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia: who? When? Why?
Biomarker Research (2020)
-
Landscape and function of multiple mutations within individual oncogenes
Nature (2020)
-
Treating hematological malignancies with drugs inhibiting ribosome biogenesis: when and why
Journal of Hematology & Oncology (2018)
