
Abstract
Background:
Platinum resistance may be attributable to inherent or acquired proficiency in homologous recombination repair (HRR) in epithelial ovarian cancer (EOC). The objective of this study was to evaluate the efficacy of the small molecule inhibitor triapine to disrupt HRR and sensitise BRCA wild-type EOC cells to platinum-based combination therapy in vitro and in vivo.
Methods:
The sensitivity of BRCA wild-type cancer cells to olaparib, cisplatin, carboplatin, doxorubicin, or etoposide in combination with triapine was evaluated by clonogenic survival assays. The effects of triapine on HRR activity in cells were measured with a DR-GFP reporter assay. The ability of triapine to enhance the effects of the carboplatin-doxil combination on EOC tumour growth delay was determined using a xenograft tumour mouse model.
Results:
Platinum resistance is associated with wild-type BRCA status. Triapine inhibits HRR activity and enhances the sensitivity of BRCA wild-type cancer cells to cisplatin, olaparib, and doxorubicin. However, sequential combination of triapine and cisplatin is necessary to achieve synergism. Moreover, triapine potentiates platinum-based combination therapy against BRCA wild-type EOC cells and produces significant delay of EOC tumour growth.
Conclusions:
Triapine promises to augment the clinical efficacy of platinum-based combination regimens for treatment of platinum-resistant EOC with wild-type BRCA and proficient HRR activity.
Similar content being viewed by others


Main
Ovarian cancer is the fifth leading cancer death in women and the most lethal gynaecologic malignancy in the United States, with more than 14 000 women dying of the disease each year and the 5-year relative survival rate is only 44% (Siegel et al, 2015). The current standard of care for patients with ovarian cancer is optimal surgical cytoreduction followed by carboplatin in combination with paclitaxel chemotherapy (du Bois et al, 2003). Ovarian cancer remains one of the most chemosensitive solid malignancies. Patients with advanced disease at time of presentation have a 65–70% chance of achieving complete clinical response after surgery and chemotherapy. However, the majority of women will experience recurrence and ultimately die of their disease due to tumour acquisition of chemoresistance (Bookman et al, 1996). Topotecan and liposomal doxorubicin (Doxil) are currently used as second-line therapy for platinum-resistant ovarian cancer (Gordon et al, 2001). Platinum-resistant women have a 10–15% chance of responding to second-line chemotherapy agents with a median overall survival of 9–10 months, and platinum refractory women have an objective response to second-line chemotherapy of <10% (Gordon et al, 2001).
Cisplatin and carboplatin are platinum-containing anticancer agents that cause predominantly intrastrand crosslinking of DNA (85–90%) responsible for their cytotoxic activity (Fichtinger-Schepman et al, 1985; Siddik, 2003). The minor interstrand crosslinks accounted for about 1% of total platination are also widely considered to be most lethal lesions caused by platinum drugs. Proficiency in the repair of platinum-DNA crosslinking lesions has a critical role in the sensitivity of cancer cells to these agents. Nucleotide excision repair (NER) functions as a primary mechanism that efficiently repairs intrastrand crosslinks (O'Donovan et al, 1994). Homologous recombination repair (HRR) can also repair single-strand gaps owing to re-initiation of leading-strand synthesis downstream of intrastrand crosslinks (Li and Heyer, 2008). In contrast, the repair of interstrand crosslinks requires NER, HRR, and translesion synthesis (TLS) acting in a coordinated manner (Nojima et al, 2005; Deans and West, 2011). Therefore, NER, HRR, and TLS contribute to platinum resistance dependent on the context of cancer types and the competency of these repair pathways (Zdraveski et al, 2000).
Hereditary breast cancer and EOC are largely attributable to germline BRCA mutations causing defects in the HRR pathway for DNA double-strand break (DSB) repair (Ratner et al, 2012). PARP inhibitors impair the process of single-strand break repair and result in DSBs. Therefore, BRCA-mutated cancer cells are hypersensitive to PARP inhibitors (Bryant et al, 2005; Farmer et al, 2005). It is estimated that about 50% of high-grade serous EOC are defective in the HRR pathway (Cancer Genome Atlas Research Network, 2011). Epithelial ovarian cancer patients with BRCA mutations show greater clinical responses to the PARP inhibitor olaparib than EOC patients without BRCA mutations (Audeh et al, 2010; Kaye et al, 2012). Given that BRCA-mutant EOC also exhibits a marked increase in sensitivity and clinical response to platinum-based drugs (Sgagias et al, 2004; Chetrit et al, 2008), it suggests that HRR is the primary mechanism for the repair of DNA damage caused by both classes of agents. Therefore, disruption of HRR can be exploited to augment the sensitivity of BRCA wild-type or HRR-proficient EOC to platinum and PARP inhibitor therapy.
Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone) is a small molecule inhibitor of ribonucleotide reductase (RNR) developed in our laboratory (Liu et al, 1992; Finch et al, 1999, 2000). Ribonucleotide reductase is a multimeric enzyme consisting of R2 and R1 subunits during the S phase of the cell cycle (Tanaka et al, 2000; Lin et al, 2004). It is responsible for the de novo synthesis of the deoxyribonucleoside diphosphates, the precursors of deoxyribonucleoside triphosphates (dNTPs) required for DNA replication and repair (Thelander and Reichard, 1979). As a potent iron chelator, triapine inactivates RNR by forming a triapine-Fe complex that quenches the tyrosyl radical at the catalytic pocket within the R2 subunit (Sartorelli et al, 1976; Popovic-Bijelic et al, 2011). Therefore, triapine effectively stalls DNA replication by inhibiting de novo biosynthesis and depletion of purine nucleotides/dNTPs in proliferating cells (Cory et al, 1995; Lin et al, 2011). We have previously demonstrated that triapine abolishes olaparib-induced BRCA1 and Rad51 foci, and disrupts BRCA1 interaction with the MRN complex. Furthermore, triapine blocks CDK2-mediated CtIP phosphorylation and DSB end resection, thereby sensitising EOC cells to PARP inhibitors (Lin et al, 2014). Preclinical and clinical studies show that triapine sensitises cancer cells to a range of DNA-damaging agents and radiation (Finch et al, 2000; Barker et al, 2006; Kunos et al, 2010). Given the clinical importance of platinum drugs in ovarian cancer treatment, the objective of this present study was to evaluate the effectiveness of triapine in the enhancement of currently used platinum-based regimens in BRCA wild-type EOC cells in vitro and in vivo.
Materials and Methods
Cell lines and chemicals
MDA-MB231, BG-1, IGROV-1, SKOV-3, A2780-CP70, PEO1, PEO4 cancer cell lines were grown in DMEM/F12 (BG-1), RPMI (MDA-MB231, IGROV-1), McCoys 5A (SKOV-3), and DMEM (A2780-CP70, PEO1, PEO4) media supplemented with 10% FBS and penicillin-streptomycin antibiotics. MDA-MB231 and SKOV-3 cells were purchased from ATCC (Manassas, VA, USA). BG-1 and IGROV-1 cells were obtained from Dr Joanne Weidhaas (Yale University); A2780-CP70 cells were provided by Dr Gil Mor (Yale University). PEO1 and PEO4 cells were provided by Dr Peter Glazer (Yale University). Cisplatin and carboplatin were purchased from EMD Millipore (Billerica, MA, USA), doxorubicin from Enzo Life Sciences (Farmingdale, NY, USA), etoposide from R&D Systems (Minneapolis, MN, USA), Doxil from Avanti Polar Lipids (Alabaster, AB, USA), and olaparib from Selleck (Houston, TX, USA), and Axon Medchem (Groningen, The Netherlands). Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone) and triapine isethionate were synthesised in our laboratory as previously described (Liu et al, 1992).
Clonogenic assays
Cells were plated at various densities in 6-well plates in triplicate. After 24 h of incubation, cells were treated with single drugs or drug combinations. After 10–21 days, colonies were fixed and stained with crystal violet solution, and counted to determine the % survival using a GelDoc imaging system with QuantityOne software (Bio-Rad, Hercules, CA, USA).
HRR assays
SKOV-3-DR-GFP cells were established as described previously (Lin et al, 2014); cells were transfected with the empty vector pcDNA-Neo or the I-SceI endonuclease expression vector pCBASceI (provided by Dr Maria Jasin, Memorial Sloan-Kettering Cancer Center) (Pierce et al, 1999) using the TransFast reagent (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Five hours after transfection, cells were treated with various concentrations of triapine for 48 h. For siRNA transfection, cells were transfected with 50 nM siRNA using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) for 16 h before transfection with pcDNA-Neo or pCBASceI plasmid and incubated for 48 h. Thereafter, cells were trypsinised and green fluorescence and side scatter of 50 000 cells were analysed by flow cytometry using an LSR II flow cytometer (BD Biosciences; San Jose, CA, USA). The cell population with an increase in GFP above the baseline level was gated to determine the percentage of GFP-positive cells. Data analyses were performed using the FlowJo software (Tree Star, Ashland, OR, USA). BRCA1, Rad51, and R2-RNR siRNAs have been described previously (Lin et al, 2004, 2014). BRCA2-siRNA (SMARTpool, siGENOME) was purchased from GE-Dharmacon (Lafayette, CO, USA).
NHEJ assays
The methodology has been described previously (Li et al, 2012). The pGL3-control plasmid (Promega) was linearised with the restriction enzyme HindIII and purified. SKOV3 cells were co-transfected with linearised pGL3-control and the internal control pRL-TK plasmids (Promega) at a 10 : 1 ratio using the TransFast reagent (Promega). After 2 h, cells were treated with various concentrations of triapine for 48 h. Cell lysates were assayed for firefly and renilla luciferase activities using the Dual Luciferase Assay System and a luminometer (Promega). The activity of firefly luciferase activity was normalised to that of renilla luciferase to correct for transfection efficiency. Overall NHEJ activity was expressed as a percentage of the luciferase activity in cells transfected with non-linearised pGL3-control plasmid. For siRNA-treated cells, cells were transfected with siRNA for 18 h before transfection with linearised pGL3-control and pRL-TK plasmids.
Combination index and statistical analyses
Combination index (CI) for evaluating the effects of two drugs in combination was performed using the CalcuSyn software (Biosoft, Cambridge, UK) based on the method of Chou and Talalay (1984). CI<1, CI=1, and CI>1 represent synergism, additivity, and antagonism, respectively. For statistical analyses, data were compared using the paired Student’s t-test. A P-value of <0.05 was considered as statistically significant.
HPLC analysis
Samples containing triapine in Tris-HCl or potassium buffer were mixed with an equal volume of CH3CN, and incubated for 10 min at room temperature then centrifuged at 10 000 g for 10 min. The samples were then analysed using an HPLC protocol, which involves two eluting buffers A (30% CH3CN, in 30 mM KPi, pH 7.4) and B (75% CH3CN, in 30 mM KPi, pH 7.4) and utilises a 5 micron 220 × 4.6 mm RP-18 C-18 reverse phase column (Applied Biosystems, Foster City, CA, USA). The flow rate was 0.6 ml per minute, starting with a mixture of 90% A and 10% B, for 5 min; this was followed by a linear gradient over the next 10 min to 100% B and continued at this composition for another 15 min before returning to the starting composition for 3 min. A UV/visible detector monitored the triapine elution at 355 nm. Triapine eluted as a sharp peak at ∼5.1 min. The sensitivity of this method was insufficient to accurately determine the concentrations of triapine that were <1 μ M.
Spectroscopic determination of the reaction of triapine and cisplatin
Reaction of triapine with cisplatin was monitored by following absorbance bleaching due to triapine loss at 355 nm versus time. The reaction kinetics was followed in 20 mM Tris-HCl buffer pH 7.0 and 37 °C with regular time interval scans. The half-reaction time was ∼20 min using 20 μ M triapine and 500 μ M cisplatin. These relatively high concentrations were used to speed the reaction and discover the fractional change in absorption upon complete reaction to enable accurate calculations under other conditions. Following an extended reaction time (100 min) with a 25-fold molar excess of cisplatin, the absorbance at 355 nm was decreased by 61% and this was assumed to represent the reaction end point. Cisplatin has no significant absorbance at 355 nm.
Tumour xenografts
The Yale University Institutional Animal Care and Use Committee approved the protocols for the in vivo animal studies in compliance with the US Public Health Policy on Humane Care and Use of Laboratory Animals. Five- to six-week-old female athymic Nude-Foxn1nu mice were purchased from Harlan Laboratories (Indianapolis, IN, USA). SKOV-3 cells suspended in 100 μl McCoy’s 5A medium mixed with 50 μl Matrigel (BD Biosciences) were implanted s.c. in the right dorsal medial area (3.6 × 106 cells per mouse). Therapy was initiated 10 days after implantation when tumours were approximately 50–100 mm3 in volume. A control group (2% DMSO) and the groups treated with triapine isethionate (dissolved in 2% DMSO, 10 mg kg−1) i.p. once daily for 5 consecutive days per week for a total of 4 weeks. Carboplatin (dissolved in water, 25 mg kg−1) and doxil (6 mg kg−1) were simultaneously given i.p. once on day 1 and day 15. For the group treated with the triple combination, mice were treated with triapine 6 h before doxil and carboplatin. Tumour size was measured three times per week using a digital caliper. Tumour volumes were calculated using the formula: length × width2/2. Body weights of mice were measured on every treatment day before administration and on the same schedule as tumour measurements during the 4-week treatment period. Tumours were excised immediately after euthanasia and fixed with 10% formalin for 72 h. Fixed tumour tissue was paraffin-embedded and sectioned for haematoxylin and eosin (H&E) staining.
Results
Cancer cell lines exhibit a profile of platinum and PARP inhibitor sensitivity correlated with BRCA status
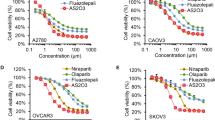
To correlate the BRCA function with platinum and PARP inhibitor sensitivity, a panel of cancer cell lines with defined BRCA status was examined for their sensitivity to cisplatin and olaparib by clonogenic assays. BG-1, MDA-MB231, CP70, SKOV-3, and PEO4 are BRCA wild-type cells. IGROV-1 is a BRCA1 heterozygous (BRCA1+/−) mutant (Foray et al, 1999) and PEO1 is a hemizygous BRCA2 mutant (Sakai et al, 2009). CP70 is a platinum-resistant EOC cell line derived from platinum-sensitive A2780 cells (Behrens et al, 1987). CP70, BG-1, and MBA-MB231 cells exhibited the highest degree of resistance to cisplatin, whereas PEO1 cells displayed the highest sensitivity to cisplatin (Figure 1A, D and E). SKOV-3, PEO4, and CP70 cells were the most resistant while PEO1 cells were the most sensitive to olaparib (Figure 1B, D and E). IGROV-1 cells manifested an intermediate sensitivity to both cisplatin and olaparib at levels between BRCA wild-type and mutant cancer cell lines (Figure 1A, B and E). All cell lines exhibited a similar sensitivity to triapine at concentrations up to 0.5 μ M, whereas PEO1, BG-1, and IGROV-1 became increasingly sensitive at 0.75 μ M (Figure 1C–E). These results suggest that platinum- and PARP inhibitor-induced DNA lesions are commonly repaired by the functional BRCA pathway that upholds resistant phenotypes of breast cancer and EOC cells.

Sensitivity profiles of breast cancer and EOC cell lines to cisplatin ( A ), olaparib ( B ), and triapine ( C). Cells were plated and treated continuously with various concentrations of the indicated agents. After 10–21 days, colonies were stained and clonogenicity was determined. Data are means±s.d. Comparison of IC50 (D) and IC90 (E) of triapine, cisplatin, and olaparib among cancer cell lines. Inhibitory concentrations of triapine, cisplatin, and olaparib at 50% and 90% of clonogenic survival (IC50 and IC90) were determined from the dose response curves shown in (A–C).
Triapine sensitises BRCA wild-type cancer cells to olaparib and low concentrations of cisplatin
Triapine has been shown to sensitise cancer cells to radiation and a variety of DNA damaging chemotherapeutic agents (Finch et al, 2000; Barker et al, 2006; Kunos et al, 2010). Given that HRR is necessary for the repair of DSBs directly or indirectly caused by these modalities, we postulate that triapine sensitises BRCA wild-type cancer cells to platinum and DNA-damaging agents by disrupting HRR. As determined by CI values for drug combinations, BG-1 cells were synergistically sensitised to the entire range of olaparib by treatment with 0.5 and 0.75 μ M triapine (Figure 2A and Supplementary Table 1A). Unexpectedly, treatment with 0.75 μ M triapine only sensitised BG-1 cells to low concentrations (0.3125 and 0.625 μ M) of cisplatin (Figure 2B and Supplementary Table 1B). Moreover, triapine progressively produced an antagonistic interaction (CI>1) with increasing concentrations of cisplatin. In an analogous manner, triapine at 0.75 μ M caused synergistic sensitisation of BG-1 cells to low concentrations of carboplatin while producing antagonism at high concentrations of carboplatin (Figure 2C and Supplementary Table 1C). In contrast, treatment with 0.75 μ M triapine sensitised BG-1 cells to most concentrations of doxorubicin (Figure 2D and Supplementary Table 1D).

Effects of triapine on the sensitivity of BG-1 cells to olaparib ( A ), cisplatin ( B ), carboplatin ( C ), and doxorubicin ( D). Cells were treated continuously with the indicated agents in combination with triapine at 0.25, 0.5, and 0.75 μ M. After 10 days, colonies were stained and clonogenicity was determined. Combination index (CI) was calculated for the effects of combinations on cell survival. Data are means±s.d. *, CI<1.
Cisplatin reacts with triapine and diminishes the effective concentration of triapine
Treatment of cancer cells with triapine was antagonistic with cisplatin and carboplatin at higher concentrations but not with olaparib or doxorubicin. Given that triapine contains three nucleophilic centres essential for its activity, we speculated that the highly electrophilic platinum ion (Pt3+) was reacting with triapine. Thus, increasing concentrations of the platinum drugs more rapidly depleted triapine in the medium thereby attenuating its activity. We first examined the stability of triapine in a cell-free buffer over a prolonged incubation period. In a neutral phosphate buffer at 37 °C, triapine alone exhibited no significant loss at the level detected by HPLC analysis over 96 h (Figure 3A). To determine whether cisplatin reduced the concentration of free triapine, the kinetics of triapine loss in the presence of various concentrations of cisplatin was determined spectroscopically. Using drug concentrations at levels similar to the range used in clonogenic assays, co-incubation of triapine and cisplatin resulted in a time- and concentration-dependent decrease in the free form of triapine in a neutral Tris buffer over a 5-h period (Figure 3B).

Chemical interaction between triapine and cisplatin in a cell-free buffer. (A) The stability of triapine in a neutral buffer. Five μ M triapine was incubated for the indicated times at 37 °C in 30 mM potassium phosphate buffer, pH 7.4. The concentration of triapine was determined by HPLC. (B) The kinetics of triapine loss in the presence of cisplatin. About 0.6 μ M triapine was incubated with various concentrations of cisplatin for the indicated times at 37 °C in 20 mM Tris-HCl buffer, pH 7.0. The concentration of triapine remaining was calculated from the absorbance at 355 nm. Effects of sequential combination of triapine and cisplatin on the sensitivity of BG-1 (C) and SKOV-3 (D) cells. Cells were pre-treated with various concentrations of cisplatin for 1 h followed by continuous exposure to indicated concentrations of triapine in the absence of cisplatin. Clonogenic survival was then determined. Data are means±s.d. *, CI<1.
Because cisplatin reduced the free concentration of triapine, we performed sequential exposure of EOC cells with cisplatin and triapine. BG-1 cells were pre-treated with various concentrations of cisplatin for 1 h, followed by exposure to 0.5 μ M triapine continuously in the absence of cisplatin. In sharp contrast to the concentration-dependent antagonistic interaction by simultaneous treatment with triapine and cisplatin (Figure 2B and Supplementary Table 1B), pre-treatment with cisplatin for 1 h before continuous triapine exposure led to synergistic sensitisation of BG-1 cells to the entire range of cisplatin concentrations (Figure 3C and Supplementary Table 2A). Likewise, SKOV-3 cells pre-treated with cisplatin for 1 h followed by exposure to 0.75 μ M triapine exhibited synergistic sensitisation to an entire range of cisplatin concentrations (Figure 3D and Supplementary Table 2B). Sequential treatment of MDA-MB231 with cisplatin and triapine also produced synergistic sensitisation, whereas concurrent treatment led to antagonism (Supplementary Figure 1).
Inhibition of RNR by triapine or siRNA suppressed HRR but not NHEJ activities
To substantiate that triapine sensitised BRCA wild-type cancer cells by inhibiting HRR, the effects of triapine on HRR activity were determined. SKOV-3-DR-GFP cells were transiently transfected with the I-SceI gene to generate a DSB in the truncated GFP gene. Transfection with the I-SceI gene effectively induced HRR activity as detected by an increase in the GFP-positive cell population. siRNA-mediated depletion of BRCA1, BRCA2, and Rad51 significantly suppressed I-SceI-induced HRR activity (Figure 4A and B). Transient transfection with BRCA2-siRNA was confirmed to sensitise SKOV-3 cells to olaparib and cisplatin (Supplementary Figure 2). Notably, depletion of the R2 subunit of RNR by siRNA considerably suppressed HRR activity. In consistent with this finding, triapine that targets R2 subunit to inhibit RNR activity also suppressed HRR activity in a concentration-dependent manner (Figure 4C and Supplementary Figure 3).

Effects of triapine on HRR and NHEJ activities in SKOV-3 cells. SKOV-3-DR-GFP cells were transfected with non-targeted control siRNA (−), BRCA1-siRNA, BRCA2-siRNA, Rad51-siRNA, or RNR-R2-siRNA (+) for 48 h. Total protein was analysed for the levels of BRCA1, BRCA2, Rad51, and RNR-R2 by western blotting. The protein levels of HSC70 were also determined as loading controls (A). Eighteen hours following siRNA transfection, cells were transfected with an empty vector (−SceI) or the I-SceI expression vector (+SceI), and were incubated for 48 h (B). Cells transfected with an empty vector (−SceI) or the I-SceI expression vector (+SceI) were incubated for 5 h and then treated with various concentrations of triapine for 48 h (C). GFP-positive population was determined and expressed as % HRR. Data are means±s.d. Eighteen hours following siRNA transfection, SKOV-3 cells were transfected with HindIII-linearised pGL3-control plasmid and incubated or 48 h (D). SKOV-3 cells transfected with HindIII-linearised pGL3-control plasmid were incubated for 2 h and then treated with various concentrations of triapine for 48 h (E). Luciferase activity was measured and expressed as %NHEJ. Data are means±s.d. Asterisks (P<0.05) indicate significant differences from control cells.
To investigate whether triapine treatment had impacts on NHEJ activity, a pGL3 luciferase-based NHEJ assay was performed. The restriction enzyme HindIII cuts DNA at a site between the promoter and coding region of luciferase gene in the pGL3-control plasmid. NHEJ rejoins the ends and restores luciferase activity in cells. SKOV-3 cells were transfected with HindIII-linearised pGL3-luciferase plasmid and subsequently treated with various concentrations of triapine. Depletion of R2-RNR, BRCA1, and BRCA2 had no effects on NHEJ (Figure 4D). Triapine did not inhibit NHEJ activity but enhance the activity marginally, though not significantly throughout, with increasing concentrations of triapine (Figure 4E).
Addition of DSB-inducing agents further augments synergistic effects of sequential combination of triapine and cisplatin
Etoposide and doxorubicin inhibit topoisomerase II, and therefore cause DSBs (Caldecott et al, 1990; Ciszewski et al, 2014). Thus, HRR is required to mitigate the lethality of these agents. Since that direct reaction between triapine and etoposide/doxorubicin is unlikely, SKOV-3 cells were simultaneously treated with triapine and these agents. Treatment with triapine led to synergistic sensitisation of SKOV-3 cells to etoposide or doxorubicin (Figure 5A and B).

Effects of doxorubicin on sequential combination of triapine and cisplatin. SKOV-3 cells were concurrently treated with 0.75 μ M triapine and various concentrations of etoposide (A) or doxorubicin (B). Clonogenic survival was determined. Data are means±s.d. *, CI<1. Cells were treated with various concentrations of cisplatin (1-h pulse treatment), doxorubicin, the cisplatin and doxorubicin combination at a fixed ratio (1000 : 1), or the cisplatin and doxorubicin combination plus 0.75 μ M triapine. Clonogenic survival was then determined. Data are means±s.d. (C) CI values for the combinations are shown in the bar graph (D).
We determined whether addition of doxorubicin further augmented the effects of sequential combination of triapine and cisplatin on BRCA wild-type EOC cells. SKOV-3 cells were pre-treated with cisplatin for 1 h and then exposed continuously to triapine and doxorubicin. The cisplatin and doxorubicin combination resulted in synergistic sensitisation of SKOV-3 cells only at the highest concentrations (Figure 5C and D). Inclusion of triapine led to enhancement of cell kill by the cisplatin–doxorubicin combination at all concentrations. Analysis of CI values confirmed that the synergism by the triple combination of triapine, cisplatin, and doxorubicin was considerably greater than the combinations of the double combination of cisplatin and doxorubicin (Figure 5D).
Triapine markedly enhances the effects of platinum-based combination therapy on tumour growth delay in EOC xenografts
To corroborate the effectiveness of the triple combination in vivo, a similar sequential combination strategy was evaluated using an EOC xenograft model. Nude mice bearing SKOV-3 tumour were treated with triapine for 6 h preceding the administration of the combination of carboplatin and doxil. Tumour sizes and mouse body weights were measured, and histological manifestations of tumour tissue were examined. Triapine treatment alone produced little effects on tumour growth delay (Figure 6A). Treatment with Doxil or carboplatin alone had moderate inhibitory effects on tumour growth (Supplementary Figure 4). The combination of carboplatin and doxil initially caused inhibition of tumour growth, but gradually lost its effects after day 28. Importantly, addition of triapine to the carboplatin–doxil combination led to a marked and significant enhancement of tumour growth delay compared with the carboplatin–doxil combination. In addition, monitoring changes in body weights showed no evident toxicity caused by drug treatment (Figure 6B). Furthermore, H&E stain of tumour tissue revealed that triapine added to the carboplatin–doxil combination resulted in shrinkage and disappearance of nuclei. In contrast, triapine and the carboplatin–doxil combination did not cause apparent changes in histological features of tumour tissue compared with that of the vehicle-treated control (Figure 6C).

Effects of triapine on the growth delay of EOC tumour by the doxil–carboplatin combination in nude mice. Nude mice bearing SKOV-3 tumour were treated as described in Materials and Methods. The mean of the tumour volumes from each treatment group (n=5) was determined. Data are means±s.d.; *P<0.05; ***P<0.001 (A). The means of body weights of mice were also shown (B). D+C, doxil+carboplatin. Representative histological images of H&E-stained tumour tissue from each treatment group are shown (C).
Discussion
HRR has been increasingly considered as an important DNA repair mechanism underlying intrinsic and acquired resistance to platinum-based chemotherapy. Hereditary EOC with deleterious BRCA mutations manifests hypersensitivity to PARP inhibitor and platinum therapy as a result of defective HRR (Ratner et al, 2012). Unfortunately, the majority of BRCA-mutated EOC eventually develops platinum resistance. It has been reported that functional restoration of HRR by secondary mutations of BRCA genes in BRCA-mutant EOC with platinum resistance occurs in patients who have previously received platinum-based chemotherapy (Sakai et al, 2008, 2009; Swisher et al, 2008; Norquist et al, 2011). Furthermore, additional novel mechanisms that restore HRR function and cause PARP inhibitor and platinum resistance have been identified, including downregulation of 53BP1 (Jaspers et al, 2013), loss of CHD4 (Guillemette et al, 2015), and stabilisation of mutant BRCA1 protein (Johnson et al, 2013). Given the importance of HRR as a target for current and future ovarian cancer treatment, our findings on the ability of triapine to suppress HRR and sensitise BRCA wild-type cancer cells represent a novel and translational approach to circumvent the problem of therapeutic resistance conferred by restored or acquired HRR proficiency.
We have previously shown that siRNA-mediated knockdown of R2-RNR reduces RNR activity and sensitises BRCA wild-type HCC116 cells to cisplatin (Lin et al, 2004, 2007). In this present study, triapine that targets R2-RNR produces synergistic sensitisation of BRCA wild-type cancer cells to olaparib, cisplatin, etoposide, and doxorubicin (Figures 2 and 5). Owing to the chemical interaction between triapine and platinum drugs, sequential treatment strategy for the combination is necessary to achieve the synergistic effects. Alternatively, identification of small molecule inhibitors that can disrupt protein–protein interactions between R1 and R2 subunits of RNR may preclude the problem of triapine–platinum interactions. Such small molecule inhibitors of RNR have been recently identified (Zhou et al, 2013).
Although NHEJ may not be impacted by triapine (Figure 4E), we cannot rule out the possibility that triapine interferes with other DNA repair mechanisms, given that NER is primarily required for platinum-DNA crosslinks. We have previously shown that reduced dNTP supply by limiting RNR activity has negative impact on the efficiency of NER and cell survival in response to platinum-induced DNA damage (Lin et al, 2004, 2007). It is conceivable that the activities of DNA polymerases involved in DNA repair, such as pol β, δ, and ɛ (Wood and Shivji, 1997), may be diminished by depletion of dNTPs due to triapine treatment. However, the effects of triapine were not limited to the combinations with platinum drugs. Triapine was also active in combination with other DNA-damaging agents, including olaparib, etoposide, doxorubicin (Figures 2 and 5), and radiation (Barker et al, 2006). In our previous studies, we have found that triapine abrogates BRCA1 foci and impairs etoposide-induced DSB end resection (Lin et al, 2014). In conjunction with the data of DR-GRP assays, triapine disrupts HRR as a major, if not only, mechanism to sensitise BRCA wild-type EOC cells to a range of DNA-damaging agents that directly or indirectly cause DSBs.
Systematic inhibition of HRR by small-molecule inhibitors like triapine raises the concern about its potential toxic effects on normal replicating cells and tissues. A large body of evidence indicates that wild-type p53 suppresses HRR activity (Mekeel et al, 1997; Bertrand et al, 2004; Gatz and Wiesmuller, 2006). Therefore, we postulate that normal tissues have restricted HRR and are less likely to be impacted by systematic inhibition of HRR by triapine. In contrast, the majority of EOC cells lacking functional p53 are expected to exhibit a greater usage of HRR for DSBs and therefore a greater susceptibility to the inhibitory effects of triapine. Our data of DR-GFP assays confirm that expression of wild-type p53 significantly suppresses I-SceI-induced HRR in SKOV3 cells (Supplementary Figure 5). In contrast, expression of mutant p53 (R175H) (Liu et al, 2010) appears to stimulate HRR although not statistically significant. SKOV3 cells are considered being p53-null as the protein level of wild-type p53 is undetectable. Furthermore, clinical studies of triapine in combination with cisplatin and radiation have shown that the half-life of plasma concentration of triapine is 2 h after a 2-h triapine (25 and 50 mg m−2) infusion and the regimens are well tolerated by patients (Kunos et al, 2010). Several strategies that impair HRR pathways, such as inhibitors of EGFR, HDAC, or HSP90, have been demonstrated to sensitise tumour cells to chemotherapy and radiation in vivo (Chernikova et al, 2012). Considering these preclinical and clinical findings, we propose that inhibition of HRR by triapine in combination with other genotoxic modalities may not inflict additional toxicity to patients if dosing schedules of triapine are properly implemented.
Following successful phase I and phase II clinical trials, Kunos and colleagues reported 3-year efficacy end points of triapine, cisplatin, and radiation combination therapy, demonstrating 4% relapse rate, 80% disease-free survival, and 82% overall survival in 24 patients with stage IB2-IIIB cervical cancer (Kunos and Sherertz, 2014). Given these promising results, triapine is currently being advanced to a phase II randomised trial with this triapine-based combination for treatment of advanced cervical and vaginal cancers. In contrast, a phase I study on platinum-resistant EOC showed that triapine co-administration with cisplatin produced a 17% partial response and 33% stable disease, with a duration of less than 6 months (Kunos et al, 2012). Cervical and vaginal cancer patients were given triapine (25 mg m−2) three times a week for 5 weeks, while EOC patients were given triapine (96 mg m−2) in a consecutive 4-day cycle for every 21 days. As impairment of HRR, rather than direct DNA damaging effects, by triapine may underlie the enhanced therapeutic efficacy of cisplatin and radiation, it is conceivable that a prolonged exposure of triapine at a lower concentration is better suited, and therefore contributes to the success of the cervical cancer trials.
The carboplatin–doxil combination is a clinical regime for recurrent and late-relapse EOC (Pujade-Lauraine et al, 2010; Gladieff et al, 2012). The concentrations of carboplatin and doxil used (25 mg kg−1 and 6 mg kg−1, respectively) in our xenograft studies were much lower than the doses given clinically (AUC 5 (equivalent to 100 mg kg−1), 30 mg m−2 (equivalent to 16 mg kg−1), respectively) to EOC patients (Pujade-Lauraine et al, 2010). Therefore, addition of triapine offers potential benefits in augmenting therapeutic efficacy and reducing toxicity of the combination of carboplatin and doxil. To avoid the potential interaction between carboplatin and triapine in vivo, we treated mice with triapine 6 h before administration with carboplatin based on the assumption that the plasma level of triapine has considerably declined at the time. Future investigation on the pharmacokinetics of triapine in mice will provide the information for determining the optimal timing of triapine and carboplatin administration. In conclusion, our findings demonstrate a proof-of-concept approach and provide a strong rationale for inclusion of triapine in platinum-based combination therapy to overcome platinum resistance and to achieve an improved clinical outcome in future EOC trials.
Change history
29 March 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376 (9737): 245–251.
Barker CA, Burgan WE, Carter DJ, Cerna D, Gius D, Hollingshead MG, Camphausen K, Tofilon PJ (2006) In vitro and in vivo radiosensitization induced by the ribonucleotide reductase inhibitor Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone). Clin Cancer Res 12 (9): 2912–2918.
Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF (1987) Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res 47 (2): 414–418.
Bertrand P, Saintigny Y, Lopez BS (2004) p53's double life: transactivation-independent repression of homologous recombination. Trends Genet 20 (6): 235–243.
Bookman MA, McGuire WP 3rd, Kilpatrick D, Keenan E, Hogan WM, Johnson SW, O'Dwyer P, Rowinsky E, Gallion HH, Ozols RF (1996) Carboplatin and paclitaxel in ovarian carcinoma: a phase I study of the Gynecologic Oncology Group. J Clin Oncol 14 (6): 1895–1902.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035): 913–917.
Caldecott K, Banks G, Jeggo P (1990) DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer Res 50 (18): 5778–5783.
Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474 (7353): 609–615.
Chernikova SB, Game JC, Brown JM (2012) Inhibiting homologous recombination for cancer therapy. Cancer Biol Ther 13 (2): 61–68.
Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S (2008) Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol 26 (1): 20–25.
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55.
Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ (2014) DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat 143 (1): 47–55.
Cory JG, Cory AH, Rappa G, Lorico A, Liu MC, Lin TS, Sartorelli AC (1995) Structure-function relationships for a new series of pyridine-2-carboxaldehyde thiosemicarbazones on ribonucleotide reductase activity and tumor cell growth in culture and in vivo. Adv Enzyme Regul 35: 55–68.
Deans AJ, West SC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11 (7): 467–480.
du Bois A, Luck HJ, Meier W, Adams HP, Mobus V, Costa S, Bauknecht T, Richter B, Warm M, Schroder W, Olbricht S, Nitz U, Jackisch C, Emons G, Wagner U, Kuhn W, Pfisterer J (2003) A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst 95 (17): 1320–1329.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 (7035): 917–921.
Fichtinger-Schepman AM, van der Veer JL, den Hartog JH, Lohman PH, Reedijk J (1985) Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry 24 (3): 707–713.
Finch RA, Liu M, Grill SP, Rose WC, Loomis R, Vasquez KM, Cheng Y, Sartorelli AC (2000) Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone): a potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem Pharmacol 59 (8): 983–991.
Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC (1999) Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of ribonucleotide reductase with antineoplastic activity. Adv Enzyme Regul 39: 3–12.
Foray N, Randrianarison V, Marot D, Perricaudet M, Lenoir G, Feunteun J (1999) Gamma-rays-induced death of human cells carrying mutations of BRCA1 or BRCA2. Oncogene 18 (51): 7334–7342.
Gatz SA, Wiesmuller L (2006) p53 in recombination and repair. Cell Death Differ 13 (6): 1003–1016.
Gladieff L, Ferrero A, De Rauglaudre G, Brown C, Vasey P, Reinthaller A, Pujade-Lauraine E, Reed N, Lorusso D, Siena S, Helland H, Elit L, Mahner S (2012) Carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in partially platinum-sensitive ovarian cancer patients: results from a subset analysis of the CALYPSO phase III trial. Ann Oncol 23 (5): 1185–1189.
Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ (2001) Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 19 (14): 3312–3322.
Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, Cantor SB (2015) Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev 29 (5): 489–494.
Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, Doroshow JH, Cranston A, Martin NM, Lau A, O'Connor MJ, Ganesan S, Borst P, Jonkers J, Rottenberg S (2013) Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 3 (1): 68–81.
Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, Chowdhury D, Wickramanayake A, Harrell MI, Liu JF, D'Andrea AD, Miron A, Swisher EM, Shapiro GI (2013) Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci USA 110 (42): 17041–17046.
Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, Amnon A, Bell-McGuinn KM, Chen LM, Friedlander M, Safra T, Vergote I, Wickens M, Lowe ES, Carmichael J, Kaufman B (2012) Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol 30 (4): 372–379.
Kunos C, Radivoyevitch T, Abdul-Karim FW, Fanning J, Abulafia O, Bonebrake AJ, Usha L (2012) Ribonucleotide reductase inhibition restores platinum-sensitivity in platinum-resistant ovarian cancer: a Gynecologic Oncology Group Study. J Transl Med 10: 79.
Kunos CA, Sherertz TM (2014) Long-term disease control with triapine-based radiochemotherapy for patients with stage IB2-IIIB cervical cancer. Front Oncol 4: 184.
Kunos CA, Waggoner S, von Gruenigen V, Eldermire E, Pink J, Dowlati A, Kinsella TJ (2010) Phase I trial of pelvic radiation, weekly cisplatin, and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) for locally advanced cervical cancer. Clin Cancer Res 16 (4): 1298–1306.
Li X, Heyer WD (2008) Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18 (1): 99–113.
Li YH, Wang X, Pan Y, Lee DH, Chowdhury D, Kimmelman AC (2012) Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS One 7 (6): e39588.
Lin ZP, Belcourt MF, Carbone R, Eaton JS, Penketh PG, Shadel GS, Cory JG, Sartorelli AC (2007) Excess ribonucleotide reductase R2 subunits coordinate the S phase checkpoint to facilitate DNA damage repair and recovery from replication stress. Biochem Pharmacol 73 (6): 760–772.
Lin ZP, Belcourt MF, Cory JG, Sartorelli AC (2004) Stable suppression of the R2 subunit of ribonucleotide reductase by R2-targeted short interference RNA sensitizes p53(-/-) HCT-116 colon cancer cells to DNA-damaging agents and ribonucleotide reductase inhibitors. J Biol Chem 279 (26): 27030–27038.
Lin ZP, Lee Y, Lin F, Belcourt MF, Li P, Cory JG, Glazer PM, Sartorelli AC (2011) Reduced level of ribonucleotide reductase R2 subunits increases dependence on homologous recombination repair of cisplatin-induced DNA damage. Mol Pharmacol 80 (6): 1000–1012.
Lin ZP, Ratner ES, Whicker ME, Lee Y, Sartorelli AC (2014) Triapine disrupts CtIP-mediated homologous recombination repair and sensitizes ovarian cancer cells to PARP and topoisomerase inhibitors. Mol Cancer Res 12 (3): 381–393.
Liu DP, Song H, Xu Y (2010) A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 29 (7): 949–956.
Liu MC, Lin TS, Sartorelli AC (1992) Synthesis and antitumor activity of amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J Med Chem 35 (20): 3672–3677.
Mekeel KL, Tang W, Kachnic LA, Luo CM, DeFrank JS, Powell SN (1997) Inactivation of p53 results in high rates of homologous recombination. Oncogene 14 (15): 1847–1857.
Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, Ishiai M, Yamamoto K, Takata M, Arakawa H, Buerstedde JM, Yamazoe M, Kawamoto T, Araki K, Takahashi JA, Hashimoto N, Takeda S, Sonoda E (2005) Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res 65 (24): 11704–11711.
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM (2011) Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol 29 (22): 3008–3015.
O'Donovan A, Davies AA, Moggs JG, West SC, Wood RD (1994) XPG endonuclease makes the 3' incision in human DNA nucleotide excision repair. Nature 371 (6496): 432–435.
Pierce AJ, Johnson RD, Thompson LH, Jasin M (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13 (20): 2633–2638.
Popovic-Bijelic A, Kowol CR, Lind ME, Luo J, Himo F, Enyedy EA, Arion VB, Graslund A (2011) Ribonucleotide reductase inhibition by metal complexes of Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): A combined experimental and theoretical study. J Inorg Biochem 105 (11): 1422–1431.
Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, Gebski V, Heywood M, Vasey PA, Volgger B, Vergote I, Pignata S, Ferrero A, Sehouli J, Lortholary A, Kristensen G, Jackisch C, Joly F, Brown C, Le Fur N, du Bois A (2010) Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol 28 (20): 3323–3329.
Ratner ES, Sartorelli AC, Lin ZP (2012) Poly (ADP-ribose) polymerase inhibitors: on the horizon of tailored and personalized therapies for epithelial ovarian cancer. Curr Opin Oncol 24: 564–571.
Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E, Taniguchi T (2009) Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res 69 (16): 6381–6386.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T (2008) Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451 (7182): 1116–1120.
Sartorelli AC, Agrawal KC, Tsiftsoglou AS, Moore EC (1976) Characterization of the biochemical mechanism of action of alpha-(N)-heterocyclic carboxaldehyde thiosemicarbazones. Adv Enzyme Regul 15: 117–139.
Sgagias MK, Wagner KU, Hamik B, Stoeger S, Spieker R, Huber LJ, Chodosh LA, Cowan KH (2004) Brca1-deficient murine mammary epithelial cells have increased sensitivity to CDDP and MMS. Cell Cycle 3 (11): 1451–1456.
Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22 (47): 7265–7279.
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65 (1): 5–29.
Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T (2008) Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 68 (8): 2581–2586.
Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y (2000) A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 404 (6773): 42–49.
Thelander L, Reichard P (1979) Reduction of ribonucleotides. Annu Rev Biochem 48: 133–158.
Wood RD, Shivji MK (1997) Which DNA polymerases are used for DNA-repair in eukaryotes? Carcinogenesis 18 (4): 605–610.
Zdraveski ZZ, Mello JA, Marinus MG, Essigmann JM (2000) Multiple pathways of recombination define cellular responses to cisplatin. Chem Biol 7 (1): 39–50.
Zhou B, Su L, Hu S, Hu W, Yip MLR, Wu J, Gaur S, Smith DL, Yuan Y-C, Synold TW, Horne D, Yen Y (2013) A small-molecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res 73 (21): 6484–6493.
Acknowledgements
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant K12HD047018 (ESR) and the ‘Discovery to Cure’ program at Yale University (ESR and ZPL). ESR and ZPL are Discovery to Cure Fellows.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ratner, E., Zhu, YL., Penketh, P. et al. Triapine potentiates platinum-based combination therapy by disruption of homologous recombination repair. Br J Cancer 114, 777–786 (2016). https://doi.org/10.1038/bjc.2016.54
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.54
