
Abstract
Background:
Genome-wide association studies (GWAS) have identified 18 loci associated with serous ovarian cancer (SOC) susceptibility but the biological mechanisms driving these findings remain poorly characterised. Germline cancer risk loci may be enriched for target genes of transcription factors (TFs) critical to somatic tumorigenesis.
Methods:
All 615 TF-target sets from the Molecular Signatures Database were evaluated using gene set enrichment analysis (GSEA) and three GWAS for SOC risk: discovery (2196 cases/4396 controls), replication (7035 cases/21 693 controls; independent from discovery), and combined (9627 cases/30 845 controls; including additional individuals).
Results:
The PAX8-target gene set was ranked 1/615 in the discovery (PGSEA<0.001; FDR=0.21), 7/615 in the replication (PGSEA=0.004; FDR=0.37), and 1/615 in the combined (PGSEA<0.001; FDR=0.21) studies. Adding other genes reported to interact with PAX8 in the literature to the PAX8-target set and applying an alternative to GSEA, interval enrichment, further confirmed this association (P=0.006). Fifteen of the 157 genes from this expanded PAX8 pathway were near eight loci associated with SOC risk at P<10−5 (including six with P<5 × 10−8). The pathway was also associated with differential gene expression after shRNA-mediated silencing of PAX8 in HeyA8 (PGSEA=0.025) and IGROV1 (PGSEA=0.004) SOC cells and several PAX8 targets near SOC risk loci demonstrated in vitro transcriptomic perturbation.
Conclusions:
Putative PAX8 target genes are enriched for common SOC risk variants. This finding from our agnostic evaluation is of particular interest given that PAX8 is well-established as a specific marker for the cell of origin of SOC.
Similar content being viewed by others


Main
Epithelial ovarian cancer (OC) is the most common cause of gynaecological cancer death in the United Kingdom (Cancer Research UK, 2016). The high mortality associated with the disease is in part because it is often diagnosed at an advanced stage and a better understanding of germline genetic predisposition to OC may eventually lead to precision screening and earlier diagnosis (Bowtell et al, 2015). Genome-wide association studies (GWAS) have so far identified 18 loci associated with susceptibility to all invasive OC or to its most common histological subtype, serous OC (SOC), that accounts for ∼70% of all cases (Song et al, 2009; Bolton et al, 2010; Goode et al, 2010; Bojesen et al, 2013; Couch et al, 2013; Permuth-Wey et al, 2013; Pharoah et al, 2013; Kuchenbaecker et al, 2015). Post-GWAS studies that integrate molecular phenotypes with GWAS findings are essential to elucidate the function of the known loci in SOC development and to unravel the potential role of loci that just fail to reach the threshold for genome-wide statistical significance (P<5 × 10−8; Freedman et al, 2011; Kar et al, 2015; Lawrenson et al, 2015).
The vast majority of single-nucleotide polymorphisms (SNPs) associated with cancer susceptibility lie in non-coding regions of the genome and so do not have any impact on protein structure and function. A growing body of evidence suggests that many inherited common risk variants instead fall into non-coding regulatory elements, such as enhancers or transcription factor (TF)-binding sites (Sur et al, 2013). Different alleles of these SNPs impact the biological activity of the regulatory elements and thus modify expression of a local (cis-acting) target gene or genes.
Expression of many TFs occur in a tissue-specific manner, and binding sites and transcriptional target genes for such lineage-specific TF drivers of cancer can be enriched at risk loci, also in a tissue-specific manner. For example, breast cancer risk SNPs are enriched for binding sites of the TFs ESR1 and FOXA1 in breast cancer cells while prostate cancer risk variants are enriched for androgen receptor-binding sites in prostate cells (Cowper-Sal lari et al, 2012; Lu et al, 2012; Jiang et al, 2013; Chen et al, 2015). However, for SOC, similar links between TFs and genetic risk have not been evaluated. This is partly because the TF-target gene networks active in SOC and SOC precursor cells are poorly characterised. Moreover, genome-wide TF-binding sites have not been profiled by chromatin immunoprecipitation combined with sequencing (ChIP-Seq) in SOC precursor and SOC tissues by initiatives such as the Encyclopedia of DNA Elements and the Nuclear Receptor Cistrome projects that enabled the corresponding studies for breast and prostate cancers (Tang et al, 2011; ENCODE Project Consortium, 2012).
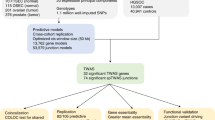
In the absence of such data, we searched for an in silico resource that would allow an agnostic evaluation of association between putative target genes of many different TFs and susceptibility to SOC. The Molecular Signatures Database (MSigDB) is a compendium of annotated functional pathways that includes 615 TF-target gene sets (Subramanian et al, 2005). All genes in each set share the same upstream cis-regulatory motif that is a predicted binding site for a particular TF and they thus represent the inferred target genes of that TF. The motifs themselves are regulatory motifs of mammalian TFs derived from the TRANSFAC database (Matys et al, 2006). In this study, we undertook pathway analysis using gene set enrichment (Subramanian et al, 2005) to test for overrepresentation of signals associated with SOC risk in these 615 TF-target gene sets using the two largest SOC GWAS data sets currently available for discovery and for independent replication. We further confirmed our top replicated gene set – targets of the TF PAX8 – using an alternative pathway analysis approach and used in vitro transcriptomic modelling to demonstrate perturbation of this gene set in the cellular context of SOC.
Materials and methods
Discovery, replication, and combined study populations
The discovery pathway analysis was performed on a meta-analysis of a North American and UK phase 1 GWAS of 2196 SOC cases and 4396 controls. The replication pathway analysis used data from 7035 SOC cases and 21 693 controls that were independent of the discovery participants and obtained from 43 case-control studies genotyped under the Collaborative Oncological Gene-environment Study (COGS) project. The two GWAS and the COGS studies have been described previously (Song et al, 2009; Permuth-Wey et al, 2011; Pharoah et al, 2013). The combined pathway analysis was based on a total of 9627 SOC cases and 30 845 controls from a meta-analysis that included the North American and UK GWAS, the COGS, and additional cases and controls from the Ovarian Cancer Association Consortium (OCAC) as reported previously (Kuchenbaecker et al, 2015). All participants were of European ancestry, provided informed consent, and had been recruited under protocols approved by a local ethics committee.
Single-nucleotide polymorphism data
The discovery, replication, and combined pathway analyses used summary findings (P-values) for association between SNP germline genotype and SOC susceptibility in the respective study populations. The discovery stage included 2 508 744 SNPs that had either been genotyped or imputed with imputation accuracy, r2>0.3 and had a minor allele frequency (MAF)>1% in both the North American and the UK GWAS. Samples were genotyped on Illumina (San Diego, CA, USA) platforms (317K/550K/610K) and imputed into the HapMap II (release 22) Utah residents with Northern and Western European ancestry (CEU) reference panel. As with most gene-based common variant association tests (Petersen et al, 2013), the gene-ranking procedure described below (Saccone et al, 2007; Christoforou et al, 2012) had been developed for HapMap-imputed GWAS and this guided our choice of HapMap-imputed SNP data over the more heavily correlated 1000 Genomes-imputed SNP data, which were also available. The replication stage was based on summary findings from COGS for a subset of 2 421 023 SNPs out of the ∼2.5 million SNPs from the discovery stage that had either been genotyped on the Illumina iCOGS custom array or imputed into the 1000 Genomes (March 2012) European reference panel with r2>0.3 and had a MAF>1% in the COGS studies. The combined pathway scan was also based on data for the same subset of SNPs but from association analysis in the combined study population. Sample and genotyping quality control, imputation, association- and meta-analysis steps for generating these three data sets have been described previously (Song et al, 2009; Permuth-Wey et al, 2011; Pharoah et al, 2013; Kuchenbaecker et al, 2015).
Gene-set enrichment analysis
Pathway analysis was conducted using the Preranked tool in the GSEA software (version 2.2.1; (Subramanian et al, 2005)) with default settings, 1000 permutations (unless otherwise specified), and no restrictions imposed on the size of gene sets that could be included. GSEA requires a list of genes ranked by any metric and a collection of annotated biological pathways or gene sets.
All 615 TF target genes sets (containing between 5 and 2657 genes; median=219 genes) annotated in the Molecular Signatures Database (MSigDB version 5.0-C3; www.broadinstitute.org/gsea/msigdb) were tested in the GSEA. Each of these gene sets represents a group of genes that share a single TF-binding site motif defined in the TRANSFAC database (version 7.4; www.gene-regulation.com; (Matys et al, 2006)). The gene sets are named after the corresponding TRANSFAC TF-binding site matrix identifier and additional details of their curation and nomenclature is available online (www.broadinstitute.org/cancer/software/gsea/wiki/index.php/MSigDB_collections#Transcription_factor_targets_.28TFT.29).
The ranked list of genes for GSEA was generated from the genome-wide SNP data by the following steps: (1) start and end positions for 23 161 genes that were unambiguously mapped were downloaded using R version 3.0.3 (Vienna, Austria; TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation package for TxDb object(s). Version 2.8.0); (2) all SNPs that lay between these start and end positions were assigned to the corresponding genes; (3) the most significant P-value among all SNPs within the boundaries of each gene was adjusted for the number of SNPs in the gene by a modification of Sidak’s correction (Saccone et al, 2007; Christoforou et al, 2012) that has been shown to reduce the effect of gene size on the P-value and account for correlations due to linkage disequilibrium between SNPs (Segrè et al, 2010); (4) the genes were ranked in descending order of the negative logarithm (base 10) of the most significant P-value (after adjustment). These steps were applied to SNPs and their P-values from the discovery (19 540 genes containing⩾1 SNP), replication, and combined data (19 364 genes containing⩾1 SNP). Quantile–quantile plots of these gene-level P-values were plotted for each data set (Supplementary Figure S1).
PAX8 target genes and pathway
The MSigDB gene set for putative targets of the TF PAX8, termed V$PAX8_B, contained 106 genes (Supplementary Table S1). These genes were grouped together because their promoter regions (±2 kb around transcription start site) contained at least one instance of the TRANSFAC motif NCNNTNNTGCRTGANNNN that matches annotation for a PAX8-binding site. Four of these genes were open reading frames and excluded from pathway analyses (Supplementary Table S1).
We used Qiagen’s Ingenuity Target Explorer (targetexplorer.ingenuity.com) to identify 55 additional genes (Supplementary Table S2) that were known to interact with PAX8 according to the literature, though not necessarily by binding PAX8 as a TF (description and citation for each interaction in Supplementary Table S3). We refer to the 157 genes (102 from MSigDB and 55 from Ingenuity) collectively as the PAX8 pathway.
PAX8 pathway analysis by interval enrichment
The INRICH tool (Lee et al, 2012) was also used to test for enrichment of genes from the PAX8 pathway within genomic intervals associated with SOC susceptibility. The 45 intervals for INRICH were generated by taking all (n=47; Supplementary Table S4) linkage-disequilibrium independent SNPs with P<10−5 for association with SOC risk in the combined data set, adding 1 Mb on either side of each SNP to capture genes potentially cis-regulated by each SNP (van Heyningen and Bickmore, 2013), and merging overlapping intervals. INRICH was used to generate 5000 sets of 45 intervals of the same width and reasonably matched to these observed intervals in terms of gene and SNP density in each interval. The number of PAX8 pathway genes overlapping the observed and permuted intervals was compared, counting multiple PAX8 pathway genes whether they overlapped a single interval, separately.
Cell culture and cell lines
IGROV1 and HeyA8 cells were cultured in Dulbecco’s Modified Eagle’s medium (Caisson, Smithfield, UT, USA) containing 10% fetal bovine serum (FBS; Seradigm, Radnor, PA, USA) and Roswell Park Memorial Institute medium (Lonza, Basel, Switzerland) containing 10% FBS, respectively. IGROV1 cells were labelled with firefly luciferase and a neomycin resistance cassette by lentiviral transduction (supernatants from Children’s Hospital Los Angeles Vector Core) and selected for by supplementing the culture media with 300 μg ml−1 G418 (Sigma-Aldrich, St Louis, MO, USA). PAX8 was silenced using individual short hairpin RNAs (shRNAs) expressed from the pLKO.1 vector (Sigma-Aldrich) and delivered by lentiviral transduction. Negative control cells were infected with a non-targeted (scrambled) hairpin. Infected clones were selected using 200 (IGROV1) and 800 (HeyA8) ng/ml puromycin (Sigma-Aldrich) and PAX8 silencing confirmed using gene expression analysis performed using TaqMan probes (Life Technologies, Carlsbad, CA, USA; Supplementary Figure S2). Cell line authentication was performed on knockdown and control lines by profiling short tandem repeats using the Promega Powerplex 16HS Assay (performed at the University of Arizona Genetics Core facility). All cultures were confirmed to be free of Mycoplasma using a Mycoplasma-specific PCR.
Microarray profiling and data analysis
RNA was extracted from the knockdown models (n=2 per cell line), cells expressing a scrambled shRNA, and parental (untreated) cells in triplicate, at independent passages. We tested five PAX8 targeting shRNAs and measured PAX8 expression levels using targeted real-time quantitative PCR performed using TaqMan gene expression probes. We then performed whole transcriptomic profiling on the samples with the greatest knockdown. Microarray profiling was performed using the Illumina HumanHT-12 v4 Expression BeadChips at the University of Southern California Epigenome Core and University of California at Los Angeles Neuroscience Genomics Core, using standard manufacturer protocols.
GenePattern (version 3.9.5; Reich et al, 2006) was used to extract signal intensity data, for cubic spline normalisation of probe expression levels (Schmid et al, 2010), and for differential expression and microarray GSEA. For differential expression, P-values from two-tailed t-tests, false discovery rate (FDR) by the Benjamini-Hochberg method, and fold changes were calculated for the following comparisons: HeyA8 untreated plus scrambled controls vs HeyA8 treated with PAX8 shRNA-1 and shRNA-2 and luciferase-labelled IGROV1 plus scrambled controls vs IGROV1 treated with PAX8 shRNA-3 and shRNA-4 (all experiments in triplicate). For microarray-GSEA, phenotype labels for these comparisons were permuted 1000 times and the standard signal-to-noise ratio was used to rank genes. Of the 157 PAX8 pathway genes, 154 were profiled on the Illumina HT-12 (three genes not profiled: LUC7L3, PKM, TMA16). Two-sided exact binomial test P-values calculated using the binom.test function in R version 3.0.3 were used to evaluate the proportion of PAX8 pathway genes that were differentially expressed.
PAX8-binding sites from chromatin immunoprecipitation with sequencing data
While we were completing our study, genome-wide maps of PAX8-binding sites compiled from ChIP-Seq of three immortalised fallopian tube secretory epithelial cell (FTSEC) lines (FT33, FT194, and FT246) and three ovarian cancer cell lines (OVSAHO, Kuramochi, and JHOS4) were published (Elias et al, 2016). We downloaded the ChIP-Seq peaks and their absolute summits called by Elias et al at FDR<0.01 from the Gene Expression Omnibus (accession number GSE79893) and defined PAX8-binding sites by extending each narrow peak to include the 500 base pair flanking sequence on either side (Heinz et al, 2010; Bailey and Machanick, 2012). We intersected SNPs at P<10−5 for association with SOC risk in the combined study population described above (n=930 genotyped or 1000 Genomes-imputed SNPs within 1 Mb of the eight unique index SNPs listed in Table 2 representing the eight intervals identified by INRICH) with these binding sites using BEDOPS version 2.4.20 (Neph et al, 2012).
Results
We began by testing the association between each of the 615 TF-target gene sets in MSigDB/TRANSFAC and SOC susceptibility by GSEA using P-values for ∼2.5 million SNPs from a meta-analysis of a North American and UK phase 1 GWAS of 2196 SOC cases and 4396 controls of European ancestry (Song et al, 2009; Permuth-Wey et al, 2011). SNPs were mapped to genes and the most significant SNP P-value for association with SOC risk in each gene was used to rank genes genome-wide for the GSEA. In this discovery pathway scan, 77 of the 615 TF-target gene sets were associated with SOC risk at PGSEA<0.05 (Supplementary Table S5) and putative target genes of the TF PAX8 (designated ‘V$PAX8_B’) emerged as the top ranked set (P<0.001; FDR=0.21; Table 1). Next, we sought to replicate these findings using genetic association results from independent samples not included in the discovery step. We performed a second GSEA for all 615 TF-target gene sets using association P-values for ∼2.4 million SNPs genome-wide in 7035 SOC cases and 21 693 controls from the Collaborative Oncological Gene-environment Study. In this replication pathway scan, 54 of the 615 TF-target gene sets were associated with SOC risk at PGSEA<0.05 (Supplementary Table S6), including 22 gene sets identified at the same significance level in the discovery analysis. The gene set containing targets of PAX8 was ranked 7th out of 615 (P=0.004; FDR=0.37; Table 1) and none of the six gene sets with a higher rank than the PAX8 targets were among the top 10 TF-target gene sets identified by the discovery step (Table 1). In order to obtain a consensus ranking of all 615 TF-target gene sets, we conducted a third GSEA using P-values for association with SOC risk from a total of 9627 SOC cases and 30 845 controls that included all discovery and replication samples and additional cases and controls from the Ovarian Cancer Association Consortium. In this combined study population, the PAX8 target gene set was once again ranked top (P<0.001; FDR=0.21; top 10 gene sets in Table 1 and full results in Supplementary Table S7).
The MSigDB/TRANSFAC PAX8 target gene set contained 102 genes (after excluding four open reading frames; Supplementary Table S1), 92 of which were covered by at least one SNP that was assessed in the genetic association studies. We expanded this gene set into what we term a PAX8 pathway by adding all genes (n=55; Supplementary Table S2) known to interact with PAX8 according to the literature, though not necessarily by binding PAX8 as a TF (description and citation for each interaction in Supplementary Table S3). This expanded 157-gene PAX8 pathway (137 of which were overlapped by at least one SNP) was also strongly associated with SOC risk in the combined study population (PGSEA=10−4 after 10 000 permutations). Next, we confirmed the association between the PAX8 pathway and SOC susceptibility using an alternative pathway analysis method called interval-based enrichment (INRICH; (Lee et al, 2012)) and used it to pinpoint specific PAX8 target genes likely driving the pathway-level signal. We identified all uncorrelated SNPs (n=47; Supplementary Table S4) associated with SOC risk at P<10−5 in the combined study population, generated two-megabase-wide intervals centred on each SNP, and merged overlapping intervals to yield 45 intervals. Fifteen of the 157 genes from the PAX8 pathway were located in eight of these 45 intervals (PINRICH=0.006 compared to 5000 permuted sets of 45 two-megabase-wide intervals). The P<10−5 index SNP at the center of five of these eight intervals was in fact genome-wide significant (P<5 × 10−8; Table 2). The SNP anchoring a sixth interval (rs2268177), though just short of genome-wide significance in the combined study population (P=9.5 × 10−7), has previously achieved this threshold in a meta-analysis of samples from OCAC with samples from the Consortium of Investigators of Modifiers of BRCA1/2 that were not included in this combined study population (Kuchenbaecker et al, 2015). Although we used a megabase flanking each P<10−5 SNP to define these intervals (to capture long-range SNP-gene cis-regulatory effects (van Heyningen and Bickmore, 2013)), in five of the eight intervals the nearest PAX8 pathway gene was less than 100 kb from the central SNP and only for two intervals did this distance extend beyond 200 kb (Table 2).
Finally, we tested whether the PAX8 pathway that had thus far been defined by combining annotations from MSigDB/TRANSFAC and curation of the published literature (that included experiments conducted in non-ovarian cell types) was cell- and cancer-type relevant. PAX8 expression was stably silenced by shRNAs in the ovarian cancer cell lines HeyA8 and IGROV1 (Supplementary Figure S2) and gene expression microarray profiling performed in knockdown and control cultures. The 157-gene PAX8 pathway, of which 154 genes were profiled on the microarray, was significantly associated with differential gene expression after PAX8 silencing in both cell line models (Pmicroarray-GSEA=0.03 for HeyA8; Pmicroarray-GSEA=0.004 for IGROV1; Supplementary Figure S3). In HeyA8 cells, 45 of these 154 genes from the PAX8 pathway were differentially expressed at P<0.05 (corresponding to a FDR<0.31; 14/154 at FDR<0.05; Pexact binomial<2.2 × 10−16 for 45/154 against 7.7/154 expected under the null hypothesis at the 5% α-level; Supplementary Table S8). In IGROV1 cells, 41 of the 154 PAX8 pathway genes were differentially expressed at P<0.05 (FDR<0.28; 17/154 at FDR<0.05; Pexact binomial<2.2 × 10−16; Supplementary Table S9). Overall, 69 of the 154 genes were differentially expressed at P<0.05 in at least one of, and 17 genes differentially expressed in both, the cell lines after PAX8 silencing (Pexact binomial=0.002 for 17/154 against 7.7/154 expected). On cross-examining results from the differential expression and INRICH analyses, we observed that of the 15 PAX8 pathway genes in eight intervals associated with SOC risk at P<10−5 (Table 2), BNC2, TNF, and NCL were differentially expressed at P<0.05 in both cell lines, HOXB5, HOXB7, HOXB8, and SP6 in IGROV1 cells only, and TERT and OSBPL7 in HeyA8 cells only (Table 3). Notably, BNC2 and HOXB7 were PAX8 target genes within 200 kb of a genome-wide significant index SNP at the 9p22.2 and 17q21.32 SOC risk loci, respectively, and differentially expressed in IGROV1 cells at FDR<0.05 (Table 3). While we were completing our study, genome-wide ChIP-Seq maps of PAX8 binding in three FTSEC and three additional ovarian cancer cell lines were published (Elias et al, 2016). We intersected these PAX8-binding sites with all SNPs at P<10−5 for association with SOC risk in the combined data set within the eight intervals identified by INRICH (+/− 1 Mb of the eight unique index SNPs listed in Table 2). SNPs at the 9p22.2, 17q21.32, and 19p13.11 genome-wide significant SOC risk loci overlapped PAX8 ChIP-binding sites in at least two of the three ovarian cancer cell lines with the most significant binding site SNP at each locus having P<7.1 × 10−8 for association with SOC risk (Supplementary Table S10).
Discussion
This is the first analysis to demonstrate that genes potentially targeted by the TF PAX8 are enriched for common genetic variation associated with SOC risk, suggesting that PAX8 may be a master transcriptional regulator of susceptibility to SOC. The emergence of the PAX8 pathway from our agnostic genome-wide approach evaluating overrepresentation of SOC risk SNPs in 615 gene sets each containing putative targets of a different TF followed by replication in independent samples and methodological replication is highly significant given recent calls for an improved understanding of the role of PAX8 in SOC biology (Bowtell et al, 2015). PAX8 encodes a member of the paired box family of TFs that contains a partial homeodomain (Chi and Epstein, 2002). It is essential for normal embryonic development of the Müllerian ducts (Mittag et al, 2007). A systematic, genome-wide RNA interference screen that included 25 ovarian cancer cell lines previously demonstrated that PAX8 is an ovarian cancer lineage-specific dependency, that is, it was the only gene that met three criteria: (a) essential for the survival and proliferation of ovarian cancer cell lines; (b) focally amplified in primary high-grade serous ovarian tumours; and (c) differentially overexpressed in ovarian cancer cell lines (Cheung et al, 2011). PAX8 has also been shown to be a key player in the proliferation, migration, and invasion of ovarian cancer cells and silencing this gene significantly inhibited anchorage-independent growth in vitro and tumour formation in a nude mouse xenograft model in vivo (Di Palma et al, 2014). Furthermore, PAX8 has been shown to drive murine SOCs originating in the fallopian tube (Perets et al, 2013). PAX8 is routinely used clinically as an epithelial marker to identify primary or metastatic tumours of Müllerian origin (Ozcan et al, 2011a, 2011b) because the TF is lineage-specific for FTSECs and ovarian surface epithelial cells (OSECs), both of which are contenders for being the cell of origin of SOC (Ozcan et al, 2011b; Adler et al, 2015).
The FDR for the PAX8-target gene set in the discovery, replication, and combined GSEA was 0.21, 0.37, and 0.21, respectively, suggesting that the result may be valid four out of five times or three out of five times. However, the convergence of orthogonal pieces of evidence: independent replication of the PAX8 GSEA findings across two of the largest genetic association studies and by INRICH, confirmation of the pathway-level association signal on including additional genes known to interact with PAX8 curated from published experiments, and significant perturbation of several of the putative PAX8 target genes near SOC risk loci in the transcriptomes of two ovarian cancer cell line models after abrogating PAX8 expression; all strongly support the observed association between this gene set and SOC risk. This convergence of evidence from integration of GWAS and cellular models also provides new insight into the potential transcriptional regulatory network of PAX8 and highlights 15 genes that are likely to be driving the association between the PAX8 pathway and risk of developing SOC (Table 3). Among these, BNC2 and HOXB7 are particularly noteworthy. BNC2 lies at 9p22.2, the first SOC risk locus to be identified (Song et al, 2009), and several functional SNPs in this region reside in enhancer elements and in a scaffold/matrix attachment region sequence that targets BNC2 (Buckley et al, under review). We have previously implicated HOXB7 at the 17q21.32 SOC risk locus in network analysis combining SOC GWAS data with SOC gene expression profiles from The Cancer Genome Atlas (Kar et al, 2015). HOXB7 overexpression in OSECs is associated with increased proliferation via fibroblast growth factor signalling (Naora et al, 2001). Our identification of SOC risk SNPs in PAX8 ChIP-binding sites at the BNC2 and HOXB7 loci further support involvement of these two genes in SOC susceptibility potentially through a PAX8-regulated mechanism. A critical next step after this pathway-based study will involve systematic genome-wide discovery of additional specific SOC risk alleles in PAX8-binding sites and/or variants that are involved in the regulation of genes that are also regulated by PAX8 (Freedman et al, 2011; Sur et al, 2013). This link between SOC risk alleles and the PAX8-target gene network may be established through dynamic expression quantitative trait locus analyses undertaken against a background of PAX8 knockdown in the relevant cell types (Califano et al, 2012; Fletcher et al, 2013).
Overall, consistent with recent pathway-level results in other cancers (Hung et al, 2015; Qian et al, 2015), the present study suggests that the genetic architecture of SOC susceptibility may be underpinned by a complex interplay between genes at the known genome-wide significant risk loci and at as yet unidentified loci that just fail to reach genome-wide significance but are functionally related to the known loci. We have demonstrated that genes interacting up- and downstream of PAX8 harbour SNPs strongly associated with SOC risk and a more comprehensive exploration of these targets may eventually open up opportunities for rational pathway-guided biomarker and therapeutic development to combat this lethal disease in its earliest stages.
Accession codes
Change history
14 February 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adler E, Mhawech-Fauceglia P, Gayther SA, Lawrenson K (2015) PAX8 expression in ovarian surface epithelial cells. Hum Pathol 46: 948–956.
Bailey TL, Machanick P (2012) Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40: e128.
Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, Edwards SL, Pickett HA, Shen HC, Smart CE, Hillman KM, Mai PL, Lawrenson K, Stutz MD, Lu Y, Karevan R, Woods N, Johnston RL, French JD, Chen X, Weischer M, Nielsen SF, Maranian MJ, Ghoussaini M, Ahmed S, Baynes C, Bolla MK, Wang Q, Dennis J, McGuffog L, Barrowdale D, Lee A, Healey S, Lush M, Tessier DC, Vincent D, Bacot F, Australian Cancer Study, Australian Ovarian Cancer Study, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab), Gene Environment Interaction and Breast Cancer (GENICA), Swedish Breast Cancer Study (SWE-BRCA), Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON), Epidemiological Study of BRCA1 & BRCA2 Mutation Carriers (EMBRACE), Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO), Vergote I, Lambrechts S, Despierre E, Risch HA, González-Neira A, Rossing MA, Pita G, Doherty JA, Alvarez N, Larson MC, Fridley BL, Schoof N, Chang-Claude J, Cicek MS, Peto J, Kalli KR, Broeks A, Armasu SM, Schmidt MK, Braaf LM, Winterhoff B, Nevanlinna H, Konecny GE, Lambrechts D, Rogmann L, Guénel P, Teoman A, Milne RL, Garcia JJ, Cox A, Shridhar V, Burwinkel B, Marme F, Hein R, Sawyer EJ, Haiman CA, Wang-Gohrke S, Andrulis IL, Moysich KB, Hopper JL, Odunsi K, Lindblom A, Giles GG, Brenner H, Simard J, Lurie G, Fasching PA, Carney ME, Radice P, Wilkens LR, Swerdlow A, Goodman MT, Brauch H, Garcia-Closas M, Hillemanns P, Winqvist R, Dürst M, Devilee P, Runnebaum I, Jakubowska A, Lubinski J, Mannermaa A, Butzow R, Bogdanova NV, Dörk T, Pelttari LM, Zheng W, Leminen A, Anton-Culver H, Bunker CH, Kristensen V, Ness RB, Muir K, Edwards R, Meindl A, Heitz F, Matsuo K, du Bois A, Wu AH, Harter P, Teo S-H, Schwaab I, Shu X-O, Blot W, Hosono S, Kang D, Nakanishi T, Hartman M, Yatabe Y, Hamann U, Karlan BY, Sangrajrang S, Kjaer SK, Gaborieau V, Jensen A, Eccles D, Høgdall E, Shen C-Y, Brown J, Woo YL, Shah M, Azmi MAN, Luben R, Omar SZ, Czene K, Vierkant RA, Nordestgaard BG, Flyger H, Vachon C, Olson JE, Wang X, Levine DA, Rudolph A, Weber RP, Flesch-Janys D, Iversen E, Nickels S, Schildkraut JM, Silva IDS, Cramer DW, Gibson L, Terry KL, Fletcher O, Vitonis AF, van der Schoot CE, Poole EM, Hogervorst FBL, Tworoger SS, Liu J, Bandera EV, Li J, Olson SH, Humphreys K, Orlow I, Blomqvist C, Rodriguez-Rodriguez L, Aittomäki K, Salvesen HB, Muranen TA, Wik E, Brouwers B, Krakstad C, Wauters E, Halle MK, Wildiers H, Kiemeney LA, Mulot C, Aben KK, Laurent-Puig P, Altena AM, Truong T, Massuger LFAG, Benitez J, Pejovic T, Perez JIA, Hoatlin M, Zamora MP, Cook LS, Balasubramanian SP, Kelemen LE, Schneeweiss A, Le ND, Sohn C, Brooks-Wilson A, Tomlinson I, Kerin MJ, Miller N, Cybulski C, Henderson BE, Menkiszak J, Schumacher F, Wentzensen N, Le Marchand L, Yang HP, Mulligan AM, Glendon G, Engelholm SA, Knight JA, Høgdall CK, Apicella C, Gore M, Tsimiklis H, Song H, Southey MC, Jager A, den Ouweland AMW, Brown R, Martens JWM, Flanagan JM, Kriege M, Paul J, Margolin S, Siddiqui N, Severi G, Whittemore AS, Baglietto L, McGuire V, Stegmaier C, Sieh W, Müller H, Arndt V, Labrèche F, Gao Y-T, Goldberg MS, Yang G, Dumont M, McLaughlin JR, Hartmann A, Ekici AB, Beckmann MW, Phelan CM, Lux MP, Permuth-Wey J, Peissel B, Sellers TA, Ficarazzi F, Barile M, Ziogas A, Ashworth A, Gentry-Maharaj A, Jones M, Ramus SJ, Orr N, Menon U, Pearce CL, Brüning T, Pike MC, Ko Y-D, Lissowska J, Figueroa J, Kupryjanczyk J, Chanock SJ, Dansonka-Mieszkowska A, Jukkola-Vuorinen A, Rzepecka IK, Pylkäs K, Bidzinski M, Kauppila S, Hollestelle A, Seynaeve C, Tollenaar RAEM, Durda K, Jaworska K, Hartikainen JM, Kosma V-M, Kataja V, Antonenkova NN, Long J, Shrubsole M, Deming-Halverson S, Lophatananon A, Siriwanarangsan P, Stewart-Brown S, Ditsch N, Lichtner P, Schmutzler RK, Ito H, Iwata H, Tajima K, Tseng C-C, Stram DO, van den Berg D, Yip CH, Ikram MK, Teh Y-C, Cai H, Lu W, Signorello LB, Cai Q, Noh D-Y, Yoo K-Y, Miao H, Iau PT-C, Teo YY, McKay J, Shapiro C, Ademuyiwa F, Fountzilas G, Hsiung C-N, Yu J-C, Hou M-F, Healey CS, Luccarini C, Peock S, Stoppa-Lyonnet D, Peterlongo P, Rebbeck TR, Piedmonte M, Singer CF, Friedman E, Thomassen M, Offit K, Hansen TVO, Neuhausen SL, Szabo CI, Blanco I, Garber J, Narod SA, Weitzel JN, Montagna M, Olah E, Godwin AK, Yannoukakos D, Goldgar DE, Caldes T, Imyanitov EN, Tihomirova L, Arun BK, Campbell I, Mensenkamp AR, van Asperen CJ, van Roozendaal KEP, Meijers-Heijboer H, Collée JM, Oosterwijk JC, Hooning MJ, Rookus MA, van der Luijt RB, Os TAM, Evans DG, Frost D, Fineberg E, Barwell J, Walker L, Kennedy MJ, Platte R, Davidson R, Ellis SD, Cole T, Bressac-de Paillerets B, Buecher B, Damiola F, Faivre L, Frenay M, Sinilnikova OM, Caron O, Giraud S, Mazoyer S, Bonadona V, Caux-Moncoutier V, Toloczko-Grabarek A, Gronwald J, Byrski T, Spurdle AB, Bonanni B, Zaffaroni D, Giannini G, Bernard L, Dolcetti R, Manoukian S, Arnold N, Engel C, Deissler H, Rhiem K, Niederacher D, Plendl H, Sutter C, Wappenschmidt B, Borg A, Melin B, Rantala J, Soller M, Nathanson KL, Domchek SM, Rodriguez GC, Salani R, Kaulich DG, Tea M-K, Paluch SS, Laitman Y, Skytte A-B, Kruse TA, Jensen UB, Robson M, Gerdes A-M, Ejlertsen B, Foretova L, Savage SA, Lester J, Soucy P, Kuchenbaecker KB, Olswold C, Cunningham JM, Slager S, Pankratz VS, Dicks E, Lakhani SR, Couch FJ, Hall P, Monteiro ANA, Gayther SA, Pharoah PDP, Reddel RR, Goode EL, Greene MH, Easton DF, Berchuck A, Antoniou AC, Chenevix-Trench G, Dunning AM (2013) Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet 45: 371–384, 384e1–e2.
Bolton KL, Tyrer J, Song H, Ramus SJ, Notaridou M, Jones C, Sher T, Gentry-Maharaj A, Wozniak E, Tsai Y-Y, Weidhaas J, Paik D, Van Den Berg DJ, Stram DO, Pearce CL, Wu AH, Brewster W, Anton-Culver H, Ziogas A, Narod SA, Levine DA, Kaye SB, Brown R, Paul J, Flanagan J, Sieh W, McGuire V, Whittemore AS, Campbell I, Gore ME, Lissowska J, Yang HP, Medrek K, Gronwald J, Lubinski J, Jakubowska A, Le ND, Cook LS, Kelemen LE, Brook-Wilson A, Massuger LFAG, Kiemeney LA, Aben KKH, van Altena AM, Houlston R, Tomlinson I, Palmieri RT, Moorman PG, Schildkraut J, Iversen ES, Phelan C, Vierkant RA, Cunningham JM, Goode EL, Fridley BL, Kruger-Kjaer S, Blaeker J, Hogdall E, Hogdall C, Gross J, Karlan BY, Ness RB, Edwards RP, Odunsi K, Moyisch KB, Baker JA, Modugno F, Heikkinenen T, Butzow R, Nevanlinna H, Leminen A, Bogdanova N, Antonenkova N, Doerk T, Hillemanns P, Dürst M, Runnebaum I, Thompson PJ, Carney ME, Goodman MT, Lurie G, Wang-Gohrke S, Hein R, Chang-Claude J, Rossing MA, Cushing-Haugen KL, Doherty J, Chen C, Rafnar T, Besenbacher S, Sulem P, Stefansson K, Birrer MJ, Terry KL, Hernandez D, Cramer DW, Vergote I, Amant F, Lambrechts D, Despierre E, Fasching PA, Beckmann MW, Thiel FC, Ekici AB, Chen X, Australian Ovarian Cancer Study Group, Australian Cancer Study (Ovarian Cancer), Ovarian Cancer Association Consortium, Johnatty SE, Webb PM, Beesley J, Chanock S, Garcia-Closas M, Sellers T, Easton DF, Berchuck A, Chenevix-Trench G, Pharoah PDP, Gayther SA (2010) Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet 42: 880–884.
Bowtell DD, Böhm S, Ahmed AA, Aspuria P-J, Bast RC, Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, Brenton JD, Chiappinelli KB, Martins FC, Coukos G, Drapkin R, Edmondson R, Fotopoulou C, Gabra H, Galon J, Gourley C, Heong V, Huntsman DG, Iwanicki M, Karlan BY, Kaye A, Lengyel E, Levine DA, Lu KH, McNeish IA, Menon U, Narod SA, Nelson BH, Nephew KP, Pharoah P, Powell DJ, Ramos P, Romero IL, Scott CL, Sood AK, Stronach EA, Balkwill FR (2015) Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 15: 668–679.
Califano A, Butte AJ, Friend S, Ideker T, Schadt E (2012) Leveraging models of cell regulation and GWAS data in integrative network-based association studies. Nat Genet 44: 841–847.
Cancer Research UK (2016) Ovarian Cancer Statistics [Online]. Available at http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/ovarian-cancer (accessed on February 2016).
Chen H, Yu H, Wang J, Zhang Z, Gao Z, Chen Z, Lu Y, Liu W, Jiang D, Zheng SL, Wei G-H, Issacs WB, Feng J, Xu J (2015) Systematic enrichment analysis of potentially functional regions for 103 prostate cancer risk-associated loci. Prostate 75: 1264–1276.
Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, Jiang G, Hsiao J, Mermel CH, Getz G, Barretina J, Gopal S, Tamayo P, Gould J, Tsherniak A, Stransky N, Luo B, Ren Y, Drapkin R, Bhatia SN, Mesirov JP, Garraway LA, Meyerson M, Lander ES, Root DE, Hahn WC (2011) Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci USA 108: 12372–12377.
Chi N, Epstein JA (2002) Getting your Pax straight: Pax proteins in development and disease. Trends Genet 18: 41–47.
Christoforou A, Dondrup M, Mattingsdal M, Mattheisen M, Giddaluru S, Nöthen MM, Rietschel M, Cichon S, Djurovic S, Andreassen OA, Jonassen I, Steen VM, Puntervoll P, Le Hellard S (2012) Linkage-disequilibrium-based binning affects the interpretation of GWASs. Am J Hum Genet 90: 727–733.
Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, Soucy P, Fredericksen Z, Barrowdale D, Dennis J, Gaudet MM, Dicks E, Kosel M, Healey S, Sinilnikova OM, Lee A, Bacot F, Vincent D, Hogervorst FBL, Peock S, Stoppa-Lyonnet D, Jakubowska A, kConFab Investigators, Radice P, Schmutzler RK, SWE-BRCA, Domchek SM, Piedmonte M, Singer CF, Friedman E, Thomassen M, Ontario Cancer Genetics Network, Hansen TVO, Neuhausen SL, Szabo CI, Blanco I, Greene MH, Karlan BY, Garber J, Phelan CM, Weitzel JN, Montagna M, Olah E, Andrulis IL, Godwin AK, Yannoukakos D, Goldgar DE, Caldes T, Nevanlinna H, Osorio A, Terry MB, Daly MB, van Rensburg EJ, Hamann U, Ramus SJ, Toland AE, Caligo MA, Olopade OI, Tung N, Claes K, Beattie MS, Southey MC, Imyanitov EN, Tischkowitz M, Janavicius R, John EM, Kwong A, Diez O, Balmaña J, Barkardottir RB, Arun BK, Rennert G, Teo S-H, Ganz PA, Campbell I, van der Hout AH, van Deurzen CHM, Seynaeve C, Gómez Garcia EB, van Leeuwen FE, Meijers-Heijboer HEJ, Gille JJP, Ausems MGEM, Blok MJ, Ligtenberg MJL, Rookus MA, Devilee P, Verhoef S, van Os TAM, Wijnen JT, HEBON, Embrace FD, Ellis S, Fineberg E, Platte R, Evans DG, Izatt L, Eeles RA, Adlard J, Eccles DM, Cook J, Brewer C, Douglas F, Hodgson S, Morrison PJ, Side LE, Donaldson A, Houghton C, Rogers MT, Dorkins H, Eason J, Gregory H, McCann E, Murray A, Calender A, Hardouin A, Berthet P, Delnatte C, Nogues C, Lasset C, Houdayer C, Leroux D, Rouleau E, Prieur F, Damiola F, Sobol H, Coupier I, Venat-Bouvet L, Castera L, Gauthier-Villars M, Léoné M, Pujol P, Mazoyer S, Bignon Y-J, GEMO Study Collaborators, Złowocka-Perłowska E, Gronwald J, Lubinski J, Durda K, Jaworska K, Huzarski T, Spurdle AB, Viel A, Peissel B, Bonanni B, Melloni G, Ottini L, Papi L, Varesco L, Tibiletti MG, Peterlongo P, Volorio S, Manoukian S, Pensotti V, Arnold N, Engel C, Deissler H, Gadzicki D, Gehrig A, Kast K, Rhiem K, Meindl A, Niederacher D, Ditsch N, Plendl H, Preisler-Adams S, Engert S, Sutter C, Varon-Mateeva R, Wappenschmidt B, Weber BHF, Arver B, Stenmark-Askmalm M, Loman N, Rosenquist R, Einbeigi Z, Nathanson KL, Rebbeck TR, Blank SV, Cohn DE, Rodriguez GC, Small L, Friedlander M, Bae-Jump VL, Fink-Retter A, Rappaport C, Gschwantler-Kaulich D, Pfeiler G, Tea M-K, Lindor NM, Kaufman B, Shimon Paluch S, Laitman Y, Skytte A-B, Gerdes A-M, Pedersen IS, Moeller ST, Kruse TA, Jensen UB, Vijai J, Sarrel K, Robson M, Kauff N, Mulligan AM, Glendon G, Ozcelik H, Ejlertsen B, Nielsen FC, Jønson L, Andersen MK, Ding YC, Steele L, Foretova L, Teulé A, Lazaro C, Brunet J, Pujana MA, Mai PL, Loud JT, Walsh C, Lester J, Orsulic S, Narod SA, Herzog J, Sand SR, Tognazzo S, Agata S, Vaszko T, Weaver J, Stavropoulou AV, Buys SS, Romero A, de la Hoya M, Aittomäki K, Muranen TA, Duran M, Chung WK, Lasa A, Dorfling CM, Miron A, BCFR, Benitez J, Senter L, Huo D, Chan SB, Sokolenko AP, Chiquette J, Tihomirova L, Friebel TM, Agnarsson BA, Lu KH, Lejbkowicz F, James PA, Hall P, Dunning AM, Tessier D, Cunningham J, Slager SL, Wang C, Hart S, Stevens K, Simard J, Pastinen T, Pankratz VS, Offit K, Easton DF, Chenevix-Trench G, Antoniou AC CIMBA (2013) Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet 9: e1003212.
Cowper-Sal lari R, Zhang X, Wright JB, Bailey SD, Cole MD, Eeckhoute J, Moore JH, Lupien M (2012) Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet 44: 1191–1198.
Di Palma T, Lucci V, de Cristofaro T, Filippone MG, Zannini M (2014) A role for PAX8 in the tumorigenic phenotype of ovarian cancer cells. BMC Cancer 14: 292.
Elias KM, Emori MM, Westerling T, Long H, Budina-Kolomets A, Li F, MacDuffie E, Davis MR, Holman A, Lawney B, Freedman ML, Quackenbush J, Brown M, Drapkin R (2016) Epigenetic remodeling regulates transcriptional changes between ovarian cancer and benign precursors. JCI Insight 1: e87988.
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74.
Fletcher MNC, Castro MAA, Wang X, de Santiago I, O’Reilly M, Chin S-F, Rueda OM, Caldas C, Ponder BAJ, Markowetz F, Meyer KB (2013) Master regulators of FGFR2 signalling and breast cancer risk. Nat Commun 4: 2464.
Freedman ML, Monteiro ANA, Gayther SA, Coetzee GA, Risch A, Plass C, Casey G, De Biasi M, Carlson C, Duggan D, James M, Liu P, Tichelaar JW, Vikis HG, You M, Mills IG (2011) Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet 43: 513–518.
Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K, Widschwendter M, Vierkant RA, Larson MC, Kjaer SK, Birrer MJ, Berchuck A, Schildkraut J, Tomlinson I, Kiemeney LA, Cook LS, Gronwald J, Garcia-Closas M, Gore ME, Campbell I, Whittemore AS, Sutphen R, Phelan C, Anton-Culver H, Pearce CL, Lambrechts D, Rossing MA, Chang-Claude J, Moysich KB, Goodman MT, Dörk T, Nevanlinna H, Ness RB, Rafnar T, Hogdall C, Hogdall E, Fridley BL, Cunningham JM, Sieh W, McGuire V, Godwin AK, Cramer DW, Hernandez D, Levine D, Lu K, Iversen ES, Palmieri RT, Houlston R, van Altena AM, Aben KKH, Massuger LFAG, Brooks-Wilson A, Kelemen LE, Le ND, Jakubowska A, Lubinski J, Medrek K, Stafford A, Easton DF, Tyrer J, Bolton KL, Harrington P, Eccles D, Chen A, Molina AN, Davila BN, Arango H, Tsai Y-Y, Chen Z, Risch HA, McLaughlin J, Narod SA, Ziogas A, Brewster W, Gentry-Maharaj A, Menon U, Wu AH, Stram DO, Pike MC, Wellcome Trust Case-Control Consortium, Beesley J, Webb PM, Australian Cancer Study (Ovarian Cancer), Australian Ovarian Cancer Study Group, Ovarian Cancer Association Consortium (OCAC), Chen X, Ekici AB, Thiel FC, Beckmann MW, Yang H, Wentzensen N, Lissowska J, Fasching PA, Despierre E, Amant F, Vergote I, Doherty J, Hein R, Wang-Gohrke S, Lurie G, Carney ME, Thompson PJ, Runnebaum I, Hillemanns P, Dürst M, Antonenkova N, Bogdanova N, Leminen A, Butzow R, Heikkinen T, Stefansson K, Sulem P, Besenbacher S, Sellers TA, Gayther SA, Pharoah PDP Ovarian Cancer Association Consortium (OCAC) (2010) A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet 42: 874–879.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589.
Hung RJ, Ulrich CM, Goode EL, Brhane Y, Muir K, Chan AT, Marchand LL, Schildkraut J, Witte JS, Eeles R, Boffetta P, Spitz MR, Poirier JG, Rider DN, Fridley BL, Chen Z, Haiman C, Schumacher F, Easton DF, Landi MT, Brennan P, Houlston R, Christiani DC, Field JK, Bickeböller H, Risch A, Kote-Jarai Z, Wiklund F, Grönberg H, Chanock S, Berndt SI, Kraft P, Lindström S, Al Olama AA, Song H, Phelan C, Wentzensen N, Peters U, Slattery ML, GECCO, Sellers TA, FOCI, Casey G, Gruber SB, CORECT, Hunter DJ, DRIVE, Amos CI, Henderson B GAME-ON Network (2015) Cross cancer genomic investigation of inflammation pathway for five common cancers: lung, ovary, prostate, breast, and colorectal cancer. J Natl Cancer Inst 107: djv246.
Jiang J, Cui W, Vongsangnak W, Hu G, Shen B (2013) Post genome-wide association studies functional characterization of prostate cancer risk loci. BMC Genomics 14 (Suppl 8): S9.
Kar SP, Tyrer JP, Li Q, Lawrenson K, Aben KKH, Anton-Culver H, Antonenkova N, Chenevix-Trench G, Australian Cancer Study, Australian Ovarian Cancer Study Group, Baker H, Bandera EV, Bean YT, Beckmann MW, Berchuck A, Bisogna M, Bjørge L, Bogdanova N, Brinton L, Brooks-Wilson A, Butzow R, Campbell I, Carty K, Chang-Claude J, Chen YA, Chen Z, Cook LS, Cramer D, Cunningham JM, Cybulski C, Dansonka-Mieszkowska A, Dennis J, Dicks E, Doherty JA, Dörk T, du Bois A, Dürst M, Eccles D, Easton DF, Edwards RP, Ekici AB, Fasching PA, Fridley BL, Gao Y-T, Gentry-Maharaj A, Giles GG, Glasspool R, Goode EL, Goodman MT, Grownwald J, Harrington P, Harter P, Hein A, Heitz F, Hildebrandt MAT, Hillemanns P, Hogdall E, Hogdall CK, Hosono S, Iversen ES, Jakubowska A, Paul J, Jensen A, Ji B-T, Karlan BY, Kjaer SK, Kelemen LE, Kellar M, Kelley J, Kiemeney LA, Krakstad C, Kupryjanczyk J, Lambrechts D, Lambrechts S, Le ND, Lee AW, Lele S, Leminen A, Lester J, Levine DA, Liang D, Lissowska J, Lu K, Lubinski J, Lundvall L, Massuger L, Matsuo K, McGuire V, McLaughlin JR, McNeish IA, Menon U, Modugno F, Moysich KB, Narod SA, Nedergaard L, Ness RB, Nevanlinna H, Odunsi K, Olson SH, Orlow I, Orsulic S, Weber RP, Pearce CL, Pejovic T, Pelttari LM, Permuth-Wey J, Phelan CM, Pike MC, Poole EM, Ramus SJ, Risch HA, Rosen B, Rossing MA, Rothstein JH, Rudolph A, Runnebaum IB, Rzepecka IK, Salvesen HB, Schildkraut JM, Schwaab I, Shu X-O, Shvetsov YB, Siddiqui N, Sieh W, Song H, Southey MC, Sucheston-Campbell LE, Tangen IL, Teo S-H, Terry KL, Thompson PJ, Timorek A, Tsai Y-Y, Tworoger SS, van Altena AM, Van Nieuwenhuysen E, Vergote I, Vierkant RA, Wang-Gohrke S, Walsh C, Wentzensen N, Whittemore AS, Wicklund KG, Wilkens LR, Woo Y-L, Wu X, Wu A, Yang H, Zheng W, Ziogas A, Sellers TA, Monteiro ANA, Freedman ML, Gayther SA, Pharoah PDP (2015) Network-based integration of gwas and gene expression identifies a hox-centric network associated with serous ovarian cancer risk. Cancer Epidemiol Biomarkers Prev 24: 1574–1584.
Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, Lawrenson K, McGuffog L, Healey S, Lee JM, Spindler TJ, Lin YG, Pejovic T, Bean Y, Li Q, Coetzee S, Hazelett D, Miron A, Southey M, Terry MB, Goldgar DE, Buys SS, Janavicius R, Dorfling CM, van Rensburg EJ, Neuhausen SL, Ding YC, Hansen TVO, Jønson L, Gerdes A-M, Ejlertsen B, Barrowdale D, Dennis J, Benitez J, Osorio A, Garcia MJ, Komenaka I, Weitzel JN, Ganschow P, Peterlongo P, Bernard L, Viel A, Bonanni B, Peissel B, Manoukian S, Radice P, Papi L, Ottini L, Fostira F, Konstantopoulou I, Garber J, Frost D, Perkins J, Platte R, Ellis S, EMBRACE, Godwin AK, Schmutzler RK, Meindl A, Engel C, Sutter C, Sinilnikova OM, GEMO Study Collaborators, Damiola F, Mazoyer S, Stoppa-Lyonnet D, Claes K, De Leeneer K, Kirk J, Rodriguez GC, Piedmonte M, O’Malley DM, de la Hoya M, Caldes T, Aittomäki K, Nevanlinna H, Collée JM, Rookus MA, Oosterwijk JC, Breast Cancer Family Registry, Tihomirova L, Tung N, Hamann U, Isaccs C, Tischkowitz M, Imyanitov EN, Caligo MA, Campbell IG, Hogervorst FBL, HEBON, Olah E, Diez O, Blanco I, Brunet J, Lazaro C, Pujana MA, Jakubowska A, Gronwald J, Lubinski J, Sukiennicki G, Barkardottir RB, Plante M, Simard J, Soucy P, Montagna M, Tognazzo S, Teixeira MR, KConFab Investigators, Pankratz VS, Wang X, Lindor N, Szabo CI, Kauff N, Vijai J, Aghajanian CA, Pfeiler G, Berger A, Singer CF, Tea M-K, Phelan CM, Greene MH, Mai PL, Rennert G, Mulligan AM, Tchatchou S, Andrulis IL, Glendon G, Toland AE, Jensen UB, Kruse TA, Thomassen M, Bojesen A, Zidan J, Friedman E, Laitman Y, Soller M, Liljegren A, Arver B, Einbeigi Z, Stenmark-Askmalm M, Olopade OI, Nussbaum RL, Rebbeck TR, Nathanson KL, Domchek SM, Lu KH, Karlan BY, Walsh C, Lester J, Australian Cancer Study (Ovarian Cancer Investigators), Australian Ovarian Cancer Study Group, Hein A, Ekici AB, Beckmann MW, Fasching PA, Lambrechts D, Van Nieuwenhuysen E, Vergote I, Lambrechts S, Dicks E, Doherty JA, Wicklund KG, Rossing MA, Rudolph A, Chang-Claude J, Wang-Gohrke S, Eilber U, Moysich KB, Odunsi K, Sucheston L, Lele S, Wilkens LR, Goodman MT, Thompson PJ, Shvetsov YB, Runnebaum IB, Dürst M, Hillemanns P, Dörk T, Antonenkova N, Bogdanova N, Leminen A, Pelttari LM, Butzow R, Modugno F, Kelley JL, Edwards RP, Ness RB, du Bois A, Heitz F, Schwaab I, Harter P, Matsuo K, Hosono S, Orsulic S, Jensen A, Kjaer SK, Hogdall E, Hasmad HN, Azmi MAN, Teo S-H, Woo Y-L, Fridley BL, Goode EL, Cunningham JM, Vierkant RA, Bruinsma F, Giles GG, Liang D, Hildebrandt MAT, Wu X, Levine DA, Bisogna M, Berchuck A, Iversen ES, Schildkraut JM, Concannon P, Weber RP, Cramer DW, Terry KL, Poole EM, Tworoger SS, Bandera EV, Orlow I, Olson SH, Krakstad C, Salvesen HB, Tangen IL, Bjorge L, van Altena AM, Aben KKH, Kiemeney LA, Massuger LFAG, Kellar M, Brooks-Wilson A, Kelemen LE, Cook LS, Le ND, Cybulski C, Yang H, Lissowska J, Brinton LA, Wentzensen N, Hogdall C, Lundvall L, Nedergaard L, Baker H, Song H, Eccles D, McNeish I, Paul J, Carty K, Siddiqui N, Glasspool R, Whittemore AS, Rothstein JH, McGuire V, Sieh W, Ji B-T, Zheng W, Shu X-O, Gao Y-T, Rosen B, Risch HA, McLaughlin JR, Narod SA, Monteiro AN, Chen A, Lin H-Y, Permuth-Wey J, Sellers TA, Tsai Y-Y, Chen Z, Ziogas A, Anton-Culver H, Gentry-Maharaj A, Menon U, Harrington P, Lee AW, Wu AH, Pearce CL, Coetzee G, Pike MC, Dansonka-Mieszkowska A, Timorek A, Rzepecka IK, Kupryjanczyk J, Freedman M, Noushmehr H, Easton DF, Offit K, Couch FJ, Gayther S, Pharoah PP, Antoniou AC, Chenevix-Trench G Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (2015) Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet 47: 164–171.
Lawrenson K, Li Q, Kar S, Seo J-H, Tyrer J, Spindler TJ, Lee J, Chen Y, Karst A, Drapkin R, Aben KKH, Anton-Culver H, Antonenkova N, Australian Ovarian Cancer Study Group, Baker H, Bandera EV, Bean Y, Beckmann MW, Berchuck A, Bisogna M, Bjorge L, Bogdanova N, Brinton LA, Brooks-Wilson A, Bruinsma F, Butzow R, Campbell IG, Carty K, Chang-Claude J, Chenevix-Trench G, Chen A, Chen Z, Cook LS, Cramer DW, Cunningham JM, Cybulski C, Dansonka-Mieszkowska A, Dennis J, Dicks E, Doherty JA, Dörk T, du Bois A, Dürst M, Eccles D, Easton DT, Edwards RP, Eilber U, Ekici AB, Fasching PA, Fridley BL, Gao Y-T, Gentry-Maharaj A, Giles GG, Glasspool R, Goode EL, Goodman MT, Grownwald J, Harrington P, Harter P, Hasmad HN, Hein A, Heitz F, Hildebrandt MAT, Hillemanns P, Hogdall E, Hogdall C, Hosono S, Iversen ES, Jakubowska A, James P, Jensen A, Ji B-T, Karlan BY, Kruger Kjaer S, Kelemen LE, Kellar M, Kelley JL, Kiemeney LA, Krakstad C, Kupryjanczyk J, Lambrechts D, Lambrechts S, Le ND, Lee AW, Lele S, Leminen A, Lester J, Levine DA, Liang D, Lissowska J, Lu K, Lubinski J, Lundvall L, Massuger LFAG, Matsuo K, McGuire V, McLaughlin JR, Nevanlinna H, McNeish I, Menon U, Modugno F, Moysich KB, Narod SA, Nedergaard L, Ness RB, Azmi MAN, Odunsi K, Olson SH, Orlow I, Orsulic S, Weber RP, Pearce CL, Pejovic T, Pelttari LM, Permuth-Wey J, Phelan CM, Pike MC, Poole EM, Ramus SJ, Risch HA, Rosen B, Rossing MA, Rothstein JH, Rudolph A, Runnebaum IB, Rzepecka IK, Salvesen HB, Schildkraut JM, Schwaab I, Sellers TA, Shu X-O, Shvetsov YB, Siddiqui N, Sieh W, Song H, Southey MC, Sucheston L, Tangen IL, Teo S-H, Terry KL, Thompson PJ, Timorek A, Tsai Y-Y, Tworoger SS, van Altena AM, Van Nieuwenhuysen E, Vergote I, Vierkant RA, Wang-Gohrke S, Walsh C, Wentzensen N, Whittemore AS, Wicklund KG, Wilkens LR, Woo Y-L, Wu X, Wu AH, Yang H, Zheng W, Ziogas A, Monteiro A, Pharoah PD, Gayther SA, Freedman ML (2015) Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat Commun 6: 8234.
Lee PH, O’Dushlaine C, Thomas B, Purcell SM (2012) INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics 28: 1797–1799.
Lu Y, Sun J, Kader AK, Kim S-T, Kim J-W, Liu W, Sun J, Lu D, Feng J, Zhu Y, Jin T, Zhang Z, Dimitrov L, Lowey J, Campbell K, Suh E, Duggan D, Carpten J, Trent JM, Gronberg H, Zheng SL, Isaacs WB, Xu J (2012) Association of prostate cancer risk with SNPs in regions containing androgen receptor binding sites captured by ChIP-On-chip analyses. The Prostate 72: 376–385.
Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110.
Mittag J, Winterhager E, Bauer K, Grümmer R (2007) Congenital hypothyroid female pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology 148: 719–725.
Naora H, Yang YQ, Montz FJ, Seidman JD, Kurman RJ, Roden RB (2001) A serologically identified tumor antigen encoded by a homeobox gene promotes growth of ovarian epithelial cells. Proc Natl Acad Sci USA 98: 4060–4065.
Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, Sandstrom R, Humbert R, Stamatoyannopoulos JA (2012) BEDOPS: high-performance genomic feature operations. Bioinformatics 28: 1919–1920.
Ozcan A, Liles N, Coffey D, Shen SS, Truong LD (2011a) PAX2 and PAX8 expression in primary and metastatic müllerian epithelial tumors: a comprehensive comparison. Am J Surg Pathol 35: 1837–1847.
Ozcan A, Shen SS, Hamilton C, Anjana K, Coffey D, Krishnan B, Truong LD (2011b) PAX 8 expression in non-neoplastic tissues, primary tumors, and metastatic tumors: a comprehensive immunohistochemical study. Mod Pathol 24: 751–764.
Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, Chen JYH, Ohman AW, Stepule CD, Kwak S, Karst AM, Hirsch MS, Setlur SR, Crum CP, Dinulescu DM, Drapkin R (2013) Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 24: 751–765.
Permuth-Wey J, Kim D, Tsai Y-Y, Lin H-Y, Chen YA, Barnholtz-Sloan J, Birrer MJ, Bloom G, Chanock SJ, Chen Z, Cramer DW, Cunningham JM, Dagne G, Ebbert-Syfrett J, Fenstermacher D, Fridley BL, Garcia-Closas M, Gayther SA, Ge W, Gentry-Maharaj A, Gonzalez-Bosquet J, Goode EL, Iversen E, Jim H, Kong W, McLaughlin J, Menon U, Monteiro ANA, Narod SA, Pharoah PDP, Phelan CM, Qu X, Ramus SJ, Risch H, Schildkraut JM, Song H, Stockwell H, Sutphen R, Terry KL, Tyrer J, Vierkant RA, Wentzensen N, Lancaster JM, Cheng JQ, Sellers TA on behalf of the Ovarian Cancer Association Consortium (OCAC) (2011) LIN28B polymorphisms influence susceptibility to epithelial ovarian cancer. Cancer Res 71: 3896–3903.
Permuth-Wey J, Lawrenson K, Shen HC, Velkova A, Tyrer JP, Chen Z, Lin H-Y, Chen YA, Tsai Y-Y, Qu X, Ramus SJ, Karevan R, Lee J, Lee N, Larson MC, Aben KK, Anton-Culver H, Antonenkova N, Antoniou AC, Armasu SM, Australian Cancer Study, Australian Ovarian Cancer Study, Bacot F, Baglietto L, Bandera EV, Barnholtz-Sloan J, Beckmann MW, Birrer MJ, Bloom G, Bogdanova N, Brinton LA, Brooks-Wilson A, Brown R, Butzow R, Cai Q, Campbell I, Chang-Claude J, Chanock S, Chenevix-Trench G, Cheng JQ, Cicek MS, Coetzee GA, Consortium of Investigators of Modifiers of BRCA1/2, Cook LS, Couch FJ, Cramer DW, Cunningham JM, Dansonka-Mieszkowska A, Despierre E, Doherty JA, Dörk T, du Bois A, Dürst M, Easton DF, Eccles D, Edwards R, Ekici AB, Fasching PA, Fenstermacher DA, Flanagan JM, Garcia-Closas M, Gentry-Maharaj A, Giles GG, Glasspool RM, Gonzalez-Bosquet J, Goodman MT, Gore M, Górski B, Gronwald J, Hall P, Halle MK, Harter P, Heitz F, Hillemanns P, Hoatlin M, Høgdall CK, Høgdall E, Hosono S, Jakubowska A, Jensen A, Jim H, Kalli KR, Karlan BY, Kaye SB, Kelemen LE, Kiemeney LA, Kikkawa F, Konecny GE, Krakstad C, Kjaer SK, Kupryjanczyk J, Lambrechts D, Lambrechts S, Lancaster JM, Le ND, Leminen A, Levine DA, Liang D, Lim BK, Lin J, Lissowska J, Lu KH, Lubiński J, Lurie G, Massuger LFAG, Matsuo K, McGuire V, McLaughlin JR, Menon U, Modugno F, Moysich KB, Nakanishi T, Narod SA, Nedergaard L, Ness RB, Nevanlinna H, Nickels S, Noushmehr H, Odunsi K, Olson SH, Orlow I, Paul J, Pearce CL, Pejovic T, Pelttari LM, Pike MC, Poole EM, Raska P, Renner SP, Risch HA, Rodriguez-Rodriguez L, Rossing MA, Rudolph A, Runnebaum IB, Rzepecka IK, Salvesen HB, Schwaab I, Severi G, Shridhar V, Shu X-O, Shvetsov YB, Sieh W, Song H, Southey MC, Spiewankiewicz B, Stram D, Sutphen R, Teo S-H, Terry KL, Tessier DC, Thompson PJ, Tworoger SS, van Altena AM, Vergote I, Vierkant RA, Vincent D, Vitonis AF, Wang-Gohrke S, Palmieri Weber R, Wentzensen N, Whittemore AS, Wik E, Wilkens LR, Winterhoff B, Woo YL, Wu AH, Xiang Y-B, Yang HP, Zheng W, Ziogas A, Zulkifli F, Phelan CM, Iversen E, Schildkraut JM, Berchuck A, Fridley BL, Goode EL, Pharoah PDP, Monteiro ANA, Sellers TA, Gayther SA (2013) Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat Commun 4: 1627.
Petersen A, Alvarez C, DeClaire S, Tintle NL (2013) Assessing methods for assigning SNPs to genes in gene-based tests of association using common variants. PLoS ONE 8: e62161.
Pharoah PDP, Tsai Y-Y, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, Buckley M, Fridley BL, Tyrer JP, Shen H, Weber R, Karevan R, Larson MC, Song H, Tessier DC, Bacot F, Vincent D, Cunningham JM, Dennis J, Dicks E, Australian Cancer Study, Australian Ovarian Cancer Study Group, Aben KK, Anton-Culver H, Antonenkova N, Armasu SM, Baglietto L, Bandera EV, Beckmann MW, Birrer MJ, Bloom G, Bogdanova N, Brenton JD, Brinton LA, Brooks-Wilson A, Brown R, Butzow R, Campbell I, Carney ME, Carvalho RS, Chang-Claude J, Chen YA, Chen Z, Chow W-H, Cicek MS, Coetzee G, Cook LS, Cramer DW, Cybulski C, Dansonka-Mieszkowska A, Despierre E, Doherty JA, Dörk T, du Bois A, Dürst M, Eccles D, Edwards R, Ekici AB, Fasching PA, Fenstermacher D, Flanagan J, Gao Y-T, Garcia-Closas M, Gentry-Maharaj A, Giles G, Gjyshi A, Gore M, Gronwald J, Guo Q, Halle MK, Harter P, Hein A, Heitz F, Hillemanns P, Hoatlin M, Høgdall E, Høgdall CK, Hosono S, Jakubowska A, Jensen A, Kalli KR, Karlan BY, Kelemen LE, Kiemeney LA, Kjaer SK, Konecny GE, Krakstad C, Kupryjanczyk J, Lambrechts D, Lambrechts S, Le ND, Lee N, Lee J, Leminen A, Lim BK, Lissowska J, Lubiński J, Lundvall L, Lurie G, Massuger LFAG, Matsuo K, McGuire V, McLaughlin JR, Menon U, Modugno F, Moysich KB, Nakanishi T, Narod SA, Ness RB, Nevanlinna H, Nickels S, Noushmehr H, Odunsi K, Olson S, Orlow I, Paul J, Pejovic T, Pelttari LM, Permuth-Wey J, Pike MC, Poole EM, Qu X, Risch HA, Rodriguez-Rodriguez L, Rossing MA, Rudolph A, Runnebaum I, Rzepecka IK, Salvesen HB, Schwaab I, Severi G, Shen H, Shridhar V, Shu X-O, Sieh W, Southey MC, Spellman P, Tajima K, Teo S-H, Terry KL, Thompson PJ, Timorek A, Tworoger SS, van Altena AM, van den Berg D, Vergote I, Vierkant RA, Vitonis AF, Wang-Gohrke S, Wentzensen N, Whittemore AS, Wik E, Winterhoff B, Woo YL, Wu AH, Yang HP, Zheng W, Ziogas A, Zulkifli F, Goodman MT, Hall P, Easton DF, Pearce CL, Berchuck A, Chenevix-Trench G, Iversen E, Monteiro ANA, Gayther SA, Schildkraut JM, Sellers TA (2013) GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet 45: 362–370 370–372.
Qian DC, Byun J, Han Y, Greene CS, Field JK, Hung RJ, Brhane Y, Mclaughlin JR, Fehringer G, Landi MT, Rosenberger A, Bickeböller H, Malhotra J, Risch A, Heinrich J, Hunter DJ, Henderson BE, Haiman CA, Schumacher FR, Eeles RA, Easton DF, Seminara D, Amos CI (2015) Identification of shared and unique susceptibility pathways among cancers of the lung, breast, and prostate from genome-wide association studies and tissue-specific protein interactions. Hum Mol Genet 24: 7406–7420.
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006) GenePattern 2.0. Nat Genet 38: 500–501.
Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PAF, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, Rice JP, Bierut LJ (2007) Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 16: 36–49.
Schmid R, Baum P, Ittrich C, Fundel-Clemens K, Huber W, Brors B, Eils R, Weith A, Mennerich D, Quast K (2010) Comparison of normalization methods for Illumina BeadChip HumanHT-12 v3. BMC Genomics 11: 349.
Segrè AV, DIAGRAM Consortium, MAGIC investigators, Groop L, Mootha VK, Daly MJ, Altshuler D (2010) Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 6: e1001058.
Song H, Ramus SJ, Tyrer J, Bolton KL, Gentry-Maharaj A, Wozniak E, Anton-Culver H, Chang-Claude J, Cramer DW, DiCioccio R, Dörk T, Goode EL, Goodman MT, Schildkraut JM, Sellers T, Baglietto L, Beckmann MW, Beesley J, Blaakaer J, Carney ME, Chanock S, Chen Z, Cunningham JM, Dicks E, Doherty JA, Dürst M, Ekici AB, Fenstermacher D, Fridley BL, Giles G, Gore ME, De Vivo I, Hillemanns P, Hogdall C, Hogdall E, Iversen ES, Jacobs IJ, Jakubowska A, Li D, Lissowska J, Lubiński J, Lurie G, McGuire V, McLaughlin J, Mędrek K, Moorman PG, Moysich K, Narod S, Phelan C, Pye C, Risch H, Runnebaum IB, Severi G, Southey M, Stram DO, Thiel FC, Terry KL, Tsai Y-Y, Tworoger SS, Van Den Berg DJ, Vierkant RA, Wang-Gohrke S, Webb PM, Wilkens LR, Wu AH, Yang H, Brewster W, Ziogas A, Houlston R, Tomlinson I, Whittemore AS, Rossing MA, Ponder BAJ, Pearce CL, Ness RB, Menon U, Kjaer SK, Gronwald J, Garcia-Closas M, Fasching PA, Easton DF, Chenevix-Trench G, Berchuck A, Pharoah PDP, Gayther SA (2009) A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet 41: 996–1000.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550.
Sur I, Tuupanen S, Whitington T, Aaltonen LA, Taipale J (2013) Lessons from functional analysis of genome-wide association studies. Cancer Res 73: 4180–4184.
Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, Liu T, Zhang Y, Brown M, Liu XS (2011) A comprehensive view of nuclear receptor cancer cistromes. Cancer Res 71: 6940–6947.
van Heyningen V, Bickmore W (2013) Regulation from a distance: long-range control of gene expression in development and disease. Philos Trans R Soc Lond B Biol Sci 368: 20120372.
Acknowledgements
This work was in part supported by a Gates Cambridge Scholarship and a Junior Research Fellowship in Clinical Medicine from Homerton College, Cambridge (to SPK) and by a K99/R00 Pathway to Independence Award (R00CA184415) from the United States National Cancer Institute (to KL). The COGS project was funded by the European Commission's Seventh Framework Programme under grant agreement HEALTH-F2-2009-223175. This project was also in part funded by the National Cancer Institute GAME-ON Post-GWAS Initiative U19-CA148112. The Ovarian Cancer Association Consortium (OCAC) is supported by a grant from the Ovarian Cancer Research Fund thanks to donations by the family and friends of Kathryn Sladek Smith (PPD/RPCI.07). Funding for the constituent studies of OCAC were provided by (these are listed by funding agency with grant numbers in parentheses): The American Cancer Society (CRTG-00-196-01-CCE); the California Cancer Research Program (00-01389V-20170, N01-CN25403, 2II0200); the Canadian Institutes for Health Research (MOP-86727); Cancer Council Victoria; Cancer Council Queensland; Cancer Council New South Wales; Cancer Council South Australia; Cancer Council Tasmania; Cancer Foundation of Western Australia; the Cancer Institute of New Jersey; Cancer Research UK (C490/A6187, C490/A10119, C490/A10124, C536/A13086, C536/A6689); the Celma Mastry Ovarian Cancer Foundation; the Danish Cancer Society (94-222-52); the ELAN Program of the University of Erlangen-Nuremberg; the Eve Appeal; the Helsinki University Central Hospital Research Fund; Helse Vest; Imperial Experimental Cancer Research Centre (C1312/A15589); the Norwegian Cancer Society; the Norwegian Research Council; the Ovarian Cancer Research Fund; Nationaal Kankerplan of Belgium; Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health Labour and Welfare of Japan; the L & S Milken Foundation; the Polish Ministry of Science and Higher Education (4 PO5C 028 14, 2 PO5A 068 27); Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Initiatives Foundation; the Roswell Park Cancer Institute Alliance Foundation; the US National Cancer Institute (K07-CA095666, K07-CA143047, K22-CA138563, N01-CN55424, N01-PC067010, N01-PC035137, P01-CA017054, P01-CA087696, P30-CA15083, P50-CA105009, P50- CA136393, R01-CA014089, R01-CA016056, R01-CA017054, R01-CA049449, R01-CA050385, R01-CA054419, R01- CA058598, R01-CA058860, R01-CA061107, R01-CA061132, R01-CA063682, R01-CA064277, R01-CA067262, R01- CA071766, R01-CA074850, R01-CA076016, R01-CA080742, R01-CA080978, R01-CA083918, R01-CA087538, R01- CA092044, R01-095023, R01-CA106414, R01-CA122443, R01-CA112523, R01-CA114343, R01-CA126841, R01- CA136924, R01-CA149429, R03-CA113148, R03-CA115195, R37-CA070867, R37-CA70867, U01-CA069417, U01- CA071966, K07-CA80668, P50-CA159981, R01CA095023, MO1- RR000056, R01-CA063678, UM1-CA186107, P01-CA87969, UM1-CA176726, UM1-CA182910, and Intramural research funds); the US Army Medical Research and Material Command (DAMD17-98-1- 8659, DAMD17-01-1-0729, DAMD17-02-1-0666, DAMD17-02-1-0669, W81XWH-10-1-02802); the National Health and Medical Research Council of Australia (199600 and 400281); the German Federal Ministry of Education and Research of Germany Programme of Clinical Biomedical Research (01 GB 9401); the state of Baden-Württemberg through Medical Faculty of the University of Ulm (P.685); the Minnesota Ovarian Cancer Alliance; the Mayo Foundation; the Fred C. and Katherine B Andersen Foundation; the Lon V. Smith Foundation (LVS-39420); the Oak Foundation; the OHSU Foundation; the Mermaid I project; the Rudolf-Bartling Foundation; the UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge, Imperial College London, the Royal Marsden Hospital; WorkSafeBC. The Nurses' Health Study and Nurses' Health Study II would like to thank the participants for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA and WY.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kar, S., Adler, E., Tyrer, J. et al. Enrichment of putative PAX8 target genes at serous epithelial ovarian cancer susceptibility loci. Br J Cancer 116, 524–535 (2017). https://doi.org/10.1038/bjc.2016.426
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.426
Keywords
This article is cited by
-
RETRACTED ARTICLE: Evaluation of PAX8 expression promotes the proliferation of stomach Cancer cells
BMC Molecular and Cell Biology (2019)
-
RETRACTED ARTICLE: Long non-coding RNA MACC1-AS1 promoted pancreatic carcinoma progression through activation of PAX8/NOTCH1 signaling pathway
Journal of Experimental & Clinical Cancer Research (2019)
-
PAX8 expression in high-grade serous ovarian cancer positively regulates attachment to ECM via Integrin β3
Cancer Cell International (2019)
-
PAX8 activates a p53-p21-dependent pro-proliferative effect in high grade serous ovarian carcinoma
Oncogene (2018)
