
Abstract
Background:
Methyl-CpG-binding domain 3 (MBD3) is an aberrant expression in human malignancies. However, the role of MBD3 in pancreatic cancer progression remains to be clarified. In this study, we investigated the effects of MBD3 on the epithelial–mesenchymal transition (EMT), and the underlying mechanism in pancreatic cancer cells.
Methods:
The abilities of migration and invasion were examined by transwell and BD Matrigel invasion assays. EMT and TGF-β/Smad signalling were evaluated.
Results:
First, we find that MBD3 expression is lower in pancreatic cancer tissues than that in non-tumour tissues, and patients with lower MBD3 levels survive significantly less than those with higher levels. Subsequently, we find that MBD3 knockdown promotes the abilities of migration and invasion, while MBD3 overexpression inhibits the above abilities. Also, MBD3 knockdown remarkably increases mesenchymal markers expression of Vimentin, α-SMA, Snail, N-cadherin, β-catenin, and downregulates epithelial markers expression of E-cadherin. On the contrary, MBD3 overexpression results in the opposite effects. Further evidence reveals that MBD3 knockdown up-regulates expression of TGF-β, and then activates p-Smad2 and p-Smad3, while MBD3 overexpression results in downregulation of TGF-β, p-Smad2, and p-Smad3.
Conclusions:
MBD3 inhibits EMT in pancreatic cancer cells probably via TGF-β/Smad signalling, and may be a new candidate target for diagnostics and prognosis of pancreatic cancer.
Similar content being viewed by others


Main
Pancreatic cancer is the fourth leading cause of cancer death in both men and women in the United States, with a 5-year survival rate limited to ∼5% (Klein, 2012). Traditional remedies such as surgery, chemotherapy, and/or radiation result in a poor clinical outcome in the past decades (Amundadottir, 2016). Lack of early diagnoses and effective interventions lead to the poor prognosis of patients (Del Chiaro et al, 2014). Therefore, it is necessary for us to look for the specific indicator and diagnosis of pancreatic cancer in the early stage.
Pancreatic cancer is characterised by early invasion and metastasis (Thota et al, 2014). It is reported that EMT has a critical role in cancer progression and metastasis (Cai et al, 2016), including pancreatic cancer (Wang et al, 2013). EMT results in losing cell adhesion and gaining migration capacity via epigenetic modifications and altered stromal interactions (Gunasinghe et al, 2012; Tam and Weinberg, 2013). Previous studies have proven that epigenetic modifications, especially DNA methylation, play an important role in pancreatic cancer (Yokoyama et al, 2016; Zagorac et al, 2016). DNA methylation controls gene expression by recruiting proteins involved in transcriptional repression (Wood and Zhou, 2016). The methyl-CpG-binding domain proteins (MBDs) read and bind methylated DNA, recruiting additional chromatin remodelling proteins that can modify histones to form compact silent chromatin (Choi et al, 2013). MBD3 is an important member of MBDs, which are important constituents of the DNA methylation machinery (Menafra and Stunnenberg, 2014). MBD3 is an essential subunit of the nucleosome remodelling and deacetylase (NuRD) complex, involved in chromatin remodelling (Zhang et al, 2016). Also, removing MBD3 allows deterministic iPSC generation (Brumbaugh and Hochedlinger, 2013). Moreover, several studies have demonstrated that MBD3 is downregulated in gastric carcinogenesis (Pontes et al, 2014), colon, and lung cancer (Zhu et al, 2004). However, the role of MBD3 in pancreatic cancer cells remains to be clarified.
In this study, we found that MBD3 expression had meaningful difference in pancreatic cancer tissues, and was critical for the ability of migration and invasion in pancreatic cancer cells in vitro. Furthermore, it was found that MBD3 inhibited EMT in pancreatic cancer cells probably via TGF-β/Smad signalling. All these findings for the first time prove that MBD3 may be a suppressor in pancreatic cancer progression.
Materials and methods
Analysis of MBD3 mRNA and protein expression in human pancreatic cancer
Correlations between pancreatic cancer histology, stage, patient survival, and MBD3 gene expression were determined through analysis of Badea and TCGA databases, which are available through Oncomine (Compendia Biosciences, www.oncomine.org) and UCSC (https://genome-cancer.ucsc.edu). High and low groups were defined as above and below the mean respectively. MBD3 protein expression in pancreatic cancer tissues and normal tissues was determined from the human protein atlas (www.proteinatlas.org).
Cell lines and cell culture
Pancreatic cancer cell lines SW1990, PaTu8988 and PANC1 were kindly provided by Second Military Medical University in Shanghai. Meanwhile, the human embryonic kidney cell line (293T) was acquired from the American Type Culture Collection. All cell lines were cultured in DMEM (Hyclone, Beijing, China) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 mg l−1 penicillin at 37 °C in a humidified incubator with 5% CO2 supply.
Real-time PCR
Total RNA was extracted using RNAiso Plus (Takara, Shiga, Japan). Reverse transcription was performed using RevertAid First Strand cDNA Synthesis Kit (Thermo, Waltham, MA, USA) according to the manufacturer’s specification. Real-time PCR was performed in triplicate in 20 μl reactions with iQ SYBR Premix Ex Taq Perfect Real Time (Bio-Rad Laboratories, Inc., Hercules, CA, USA), 50 ng first strand cDNA and 0.2 μg each primer. The primer pair used for the amplification of the human MBD3 gene was as follows: forward primer, 5′-CGGCCACAGGGATGTCTTTT-3′, and reverse primer, 5′-TGCTGGGGTGGTTGGTAATC-3′. MMP2 primer: Forward, 5′-CACAGGAGG AGAAGGCTGTG-3′ and reverse, 5′-GAGCTTGGGAAAGCCAGGAT-3′; MMP9 primer: Forward, 5′-TTCAGGGAGACGCCCATTTC-3′ and reverse, 5′-TGTAGAGTCTCTCGCTGGGG-3′; and GAPDH primer: Forward, 5′-GGTGAAGGTCGGTGTGAACG-3′ and reverse, 5′-CTCGCTCCTGGAAGATGGTG-3′. Samples were cycled once at 95 °C for 2 min, and then subjected to 35 cycles of 95 °C, 56 °C, and 72 °C for 30 s each. The relative mRNA content was calculated using the 2−ΔΔCT method with GAPDH as an endogenous control.
Western blotting
The cultured cells were rinsed with cold PBS before treated with RIPA lysis buffer at 4 °C for 10 min. Then the mixture was heated at 100 °C for 10 min and centrifuged under 4 °C at 14000 g min−1 for 10 min. The supernatant was removed, and the protein concentration was measured with the BCA method. About 20 μg of protein was loaded in each lane, separated by 10% SDS–PAGE and transferred to the PVDF membrane. The membrane was blocked with 5% non-fat milk powder for 1 h at room temperature before overnight incubation with primary antibodies 4 °C, followed by the secondary antibody. The antibodies were mouse anti-β-tubulin (Cell Signaling, Danvers, MA, USA; CAT 6181), rabbit anti-MBD3 (Cell Signaling, CAT 3896), mouse anti-Flag (Sigma, San Francisco, CA, USA; CAT F1804), rabbit anti-MMP2 (ImmunoWay, Plano, TX, USA; CAT YT2798), rabbit anti-MMP9 (ImmunoWay CAT YT1892), rabbit anti-Vimentin (Cell Signaling, CAT 5741), rabbit anti-N-cadherin (Cell Signaling, CAT 13116), rabbit anti-E-cadherin (Cell Signaling, CAT 3195), rabbit anti-β-catenin (Cell Signaling, CAT 8480), rabbit anti-Snail (Cell Signaling, CAT 3879), rabbit anti-α-SMA (Cell Signaling, CAT 14968), rabbit anti-Smad2/3 (Cell Signaling, CAT 8685), rabbit anti-P-Smad2 (Cell Signaling, CAT 3108), rabbit anti-P-Smad3 (Cell Signaling, CAT 9520), rabbit anti-TGF-β (Cell Signaling, CAT 3709).
Plasmid construction
The complete MBD3 sequence was amplified by RT–PCR using primers MBD3-all-F (5′-CGGAATTCCGATGGAGCGGAAGAGCCCGAGCG-3′) and MBD3-all-R (5′-GGGGTACCCCCTAGACGTGCTCCATCTCCGGGT-3′) from a cDNA library of PANC1 cells, then inserted into the expression Vector p3xFLAG-Myc-CMV-24 (Sigma). The MBD3 and EGFP shRNA oligos (MBD3-shRNA-F 5′-CCGGCGGCCTGAACGCCTTCGACATCTCGAGATGT CGAAGGCGTTCAGGCCGTTTTTG-3′, MBD3-shRNA-R 5′-AATTCAAAAACGGCCTGAACGCCTTCGACAT CTCGAGATGTCGAAGGCGTTCAGGCCG-3′, EGFP-shRNA-F 5′-CCGGTACAACAGCCACAACGTCTATCTCGAGATAGACGTTGTGGCTGTTGTATTTTTG-3′, EGFP-shRNA-R 5′-AATTCAAAAATACAACAGCCACAACGTCTATCTCGAGATAGACGTTGTGGCTGTTGTA-3′) were first annealed into double strands and then cloned into pLKO.1-puro (Sigma).
Transfection and infection
Vector, Flag-MBD3, sh-EGFP, and sh-MBD3 were transfected into pancreatic cancer cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The methods of generating retroviruses encoding reprogramming factors and further infection of NPCs were referenced by previous papers (Pontes et al, 2014).
Transwell migration and invasion assay
Transwell assays was carried out using transwell inserts (Corning, Corning, NY, USA) containing 8-m permeable pores according to the manufacturer’s protocol. Transfected PANC1, PaTu8988, and SW1990 cells were collected, resuspended in serum-free medium and transferred to 8-m permeable pores (∼100 000 cells per well). The chambers were then incubated for 24 h in culture medium with 10% FBS in the bottom chambers before examination. The cells on the upper surface were scraped and washed away, whereas the migrated cells on the lower surface were fixed and stained with 0.05% crystal violet for 30 min. Finally, five independent fields were counted for each transwell and the average number of cells per field was represented in the graphs. For assessing cell invasion, 100 000 cells in serum-free medium were seeded in the matrigel-coated transwell inserts (BD Bioscience, Corning, NY, USA). The cells were then processed similar to that of cell migration assay.
Statistical analyses
All data are presented as mean±s.d. from at least three independent experiments. Comparisons between groups were analysed using the Student’s t-test (two groups) or an one-way ANOVA (multiple groups). Kaplan–Meier survival was analysed using log-rank analysis. P<0.05 was considered statistically significant.
Results
MBD3 is downregulated in pancreatic cancer and associated with patients’ overall survival
To confirm the clinical relevance of MBD3 expression, we first analysed the MBD3 protein expression in clinical specimens from the human protein atlas (www.proteinatlas.org). We found that MBD3 had the negative weak expression in pancreatic cancer, and positive strong expression in normal pancreas (Figure 1A). Consistently, MBD3 mRNA level was lower in pancreatic cancer tissues than that in normal pancreatic tissues (1.354±0.0277 vs 1.687±0.04863, P<0.001, n=78) in Badea pancreas database (Badea et al, 2008; Figure 1B). Due to the limitations of the Badea pancreas database information, we investigated more information in TCGA database (https://genome-cancer.ucsc.edu), and evaluated the correlation of MBD3 expression with patients’ outcome, clinicopathological features and tumour stage. Subsequently, Patients with high expression of MBD3 had a median survival of 15 months as compared with 10 months for the patients with low expression of MBD3 (HR=1.659, P<0.05; Figure 1C). Also, MBD3 expression in pancreas adenocarcinoma ductal was lower than that in pancreas adenocarcinoma other subtype and pancreas colloid (mucinous non-cystic) carcinoma (Figure 1D). Moreover, further analysis showed that MBD3 expression decreased successively in stage I, stage II, stage III, and stage IV groups, and the stage I group presented the highest and stage IV the lowest MBD3 expression levels (Figure 1E). All these data suggest that MBD3 is downregulated in pancreatic cancer and associated with the poor outcome of patients.

MBD3 is downregulated in pancreatic cancer and associated with patients’ overall survival. (A) MBD3 protein expression in pancreatic cancer tissues and normal pancreatic tissues was analysed through the human protein atlas (www.proteinatlas.org). Magnification, × 4; bars, 500 μm. Magnification, × 40; bars, 100 μm. (B) Analysis of MBD3 mRNA levels in 78 pairs of pancreatic cancer and non-tumour tissues in Badea pancreas database. N=39 for non-tumour group, and N=39 for tumour group. ***P<0.001. (C) Analysis of the TCGA database indicates that MBD3 expression is correlated with patient’ overall survival. N=49 for MBD3-low group, and N=32 for MBD3-high group. P=0.034 was determined by Log-Rank test. (D) Analysis of the TCGA database indicates MBD3 correlates with clinicopathological features. The results are presented by heat map (left panel) and boxplot (right panel). N=139 for pancreas adenocarcinoma ductal type group, N=23 for pancreas adenocarcinoma other subtype group, and N=4 for pancreas colloid (mucinous non-cystic) carcinoma group. ***P<0.001. (E) Analysis of the TCGA database indicates MBD3 is associated with stage in pancreatic cancer. The results are presented by heat map (left panel) and boxplot (right panel). N=20 for stage I group, N=140 for stage II group, and N=4 for stage III group, and N=3 for stage IV group. *P<0.05.
MBD3 expression is profiled in pancreatic cancer cells
To further investigate the roles of MBD3 in pancreatic cancer, we first examined the expression level of MBD3 in SW1990, PaTu8988 and PANC1 cells using real-time PCR and western blot. We noticed that MBD3 expression, at both mRNA and protein levels, was higher in PANC1 cells than that in SW1990 and PaTu8988 cells (Figure 2A). Previous studies identify that SW1990 and PaTu8988 cells are derived from metastatic site: spleen (Kyriazis et al, 1983) and liver (Elsasser et al, 1992), while PANC1 cells are derived from primary site (Lieber et al, 1975), implying that MBD3 down-regulation might be involved in metastasis of pancreatic cancer cells. As MBD3 expression is relatively lower in SW1990 cells, it was used to normalise the other two cell lines as a reference. Subsequently, we constructed sh-MBD3 and Flag-MBD3 plasmids to investigate the roles of MBD3 in pancreatic cancer, sh-EGFP or Vector as a control, respectively. After transfection, the mRNA and protein levels of MBD3 significantly reduced in sh-MBD3 group compared with sh-EGFP group (Figure 2B). Vector or Flag-MBD3 was transferred into PaTu8988 and SW1990 cells, and then MBD3 overexpression was confirmed at mRNA and protein levels by real-time PCR and western blot (Figure 2C).

MBD3 expression is profiled in pancreatic cancer cells. (A) Relative expression levels of MBD3 protein and mRNA were assessed in SW1990, PaTu8988, and PANC1 cells. *P<0.05. (B) MBD3 protein and mRNA levels were reduced in sh-MBD3- PANC1 and PaTu8988 cells. **P<0.01. (C) MBD3 protein and mRNA levels were increased in PaTu8988 and SW1990 cells transfected with Flag-MBD3. ***P<0.001.
MBD3 inhibits the migration ability of pancreatic cancer cells
Next, we examined the ability of migration by transwell assays in pancreatic cancer cells. We found that the numbers of migrated cells were 108±7 and 297±11 in sh-EGFP and sh-MBD3 PANC1 cells, and 77±8 and 236±10 in sh-EGFP and sh-MBD3 PaTu8988 cells, respectively (Figure 3A and B), indicating that MBD3 knockdown improved the ability of migration in PANC1 and PaTu8988 cells. To confirm the above results, we assessed the ability of migration in PaTu8988 cells or SW1990 cells transfected with Vector or Flag-MBD3 plasmids. The numbers of migrated cells were 107±9 and 36±5 in Vector and Flag-MBD3 PaTu8988 cells, and 184±12 and 74±8 in Vector and Flag-MBD3 SW1990 cells, respectively (Figure 3C and D), indicating that MBD3 overexpression inhibited the ability of migration in pancreatic cancer cells. All these data suggest that MBD3 inhibits the ability of migration in pancreatic cancer cells.

MBD3 inhibits the migration ability of pancreatic cancer cells. (A) The ability of migration was examined using transwell assay in PANC1 and PaTu8988 cells transfected with sh-MBD3 or sh-EGFP plasmids. Representative images of migrated cells were shown. Magnification, × 20; bars, 50 μm. (B) The graph indicated the average number of migrated cells per field. **P<0.001. (C) The ability of migration was examined using transwell assay in SW1990 and PaTu8988 cells transfected with Flag-MBD3 or Vector plasmids. Representative images of migrated cells were shown. Magnification, × 20; bars, 50 μm. (D) Migrated cells were counted and analysed. *P<0.05, **P<0.01.
MBD3 suppresses the invasion ability of pancreatic cancer cells
Subsequently, we examined the ability of invasion by BD Matrigel invasion assays. PANC1 and PaTu8988 cells were transfected with sh-EGFP or sh-MBD3 plasmids, and PaTu8988 and SW1990 cells with Vector or Flag-MBD3 for 72 h. The numbers of invasive cells were 92±7 and 180±11 in sh-EGFP and sh-MBD3 PANC1 cells, and 30±6 and 80±8 in sh-EGFP and sh-MBD3 PaTu8988 cells, respectively (Figure 4A and B), indicating that knockdown of MBD3 obviously enhanced the invasion ability of PANC1 and PaTu8988 cells. Meanwhile, the numbers of invasive cells were 70±9 and 26±5 in Vector and Flag-MBD3 PaTu8988 cells, and 157±11 and 63±8 in Vector and Flag-MBD3 SW1990 cells, respectively. It is suggested that upregulation of MBD3 significantly inhibited the invasion ability of PaTu8988 and SW1990 cells (Figure 5A and B). Also, MBD3 knockdown led to upregulation of MMP2 and MMP9 at both mRNA and protein levels in PANC1 and PaTu8988 cells (Figure 4C and D), while MBD3 overexpression downregulated the expression of MMP2 and MMP9 in SW1990 and PaTu8988 cells (Figure 5C and D). Above all, data suggest that MBD3 suppresses the invasion ability of pancreatic cancer cells.

MBD3 enhances the invasion ability of pancreatic cancer cells. (A) The invasion ability was examined using BD Matrigel invasion assay in PaTu8988 and PANC1 cells transfected with sh-EGFP or sh-MBD3 plasmids. Magnification, × 20; bars, 50 μm. (B) Invasive cells were counted and analysed. **P<0.01. MMP2 and MMP9 were identified using western blotting (C) and real-time PCR (D) in above cells. *P<0.05, **P<0.01.

Up-regulating MBD3 inhibits the ability of invasion in pancreatic cancer cells. (A) The invasion ability was examined using BD Matrigel invasion assay in PaTu8988 and SW1990 cells transfected with Vector or Flag-MBD3 plasmids. Magnification, × 20; bars, 50 μm. (B) Invasive cells were counted and analysed. **P<0.01, ***P<0.001. MMP2 and MMP9 were determined using western blotting (C) and real-time PCR (D) in above cells. *P<0.05, **P<0.01.
MBD3 inhibits EMT through suppressing TGF-β/Smad signalling in pancreatic cancer cells
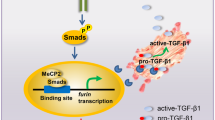
The EMT is deemed to be associated with the ability of migration and invasion in cancer cells (Karaczyn et al, 2016). Therefore, we detected the EMT markers at protein levels using western blotting. Our data suggested that MBD3 knockdown resulted in upregulation of Vimentin, α-SMA, Snail, N-cadherin, β-catenin, and downregulation of E-cadherin in PANC1 and PaTu8988 cells (Figure 6A). In contrast, MBD3 overexpression led to downregulation of Vimentin, α-SMA, Snail, N-cadherin, β-catenin, and upregulation of E-cadherin in PaTu8988 and SW1990 cells (Figure 6B). It is confirmed that MBD3 inhibits EMT in pancreatic cancer cells. As TGF-β/Smad signalling is closely related to EMT in cancer (Yoshida et al, 2016), we investigated the effects of MBD3 on the classic TGF-β/Smad signalling. We found that MBD3 knockdown upregulated TGF-β, p-Smad2/3 in PANC1 and PaTu8988 cells (Figure 6C), while MBD3 overexpression resulted in down-regulation of TGF-β, p-Smad2/3 (Figure 6D). Knocking-down or overexpressing MBD3 had no influence on total smad2/3 protein. These data suggest that MBD3 inhibits EMT in pancreatic cancer cells probably via TGF-β/Smad signalling.

MBD3 inhibits EMT through suppressing TGF- β /Smad signalling in pancreatic cancer cells. (A) MBD3 knockdown in PANC1 and PaTu8988 cells induced EMT, as detected by a decrease in E-cadherin and increases in Vimentin, α-SMA, Snail, N-cadherin and β-catenin. (B) Treatment of PaTu8988 and SW1990 cells with Flag-MBD3 reversed EMT, as defined by an increase in E-cadherin and decreases in Vimentin, α-SMA, Snail, N-cadherin and β-catenin. (C) Immunoblotting of Smad2/3, p-Smad2/3, TGF-β in PANC1 and PaTu8988 cells treated with sh-MBD3 or sh-EGFP. β-Tubulin was used as a loading control. (D) Immunoblotting of Smad2/3, p-Smad2/3, TGF-β in SW1990, and PaTu8988 cells transfected with Flag-MBD3 or Vector. β-tubulin was used as a loading control.
Discussion
In this study, we find that MBD3 may function as an anti-oncogene in pancreatic cancer. We for the first time find that MBD3 suppresses EMT in pancreatic cancer cells probably via TGF-β/Smad signalling. Importantly, we confirm that MBD3 is downregulated in human pancreatic cancer at both mRNA and protein levels, and acts as an independent prognostic factor for patient outcome. All these findings suggest that MBD3 may be a candidate for diagnosis and prognosis of pancreatic cancer.
Our previous study focused on examining the activity of MBD3 binding to hypermethylated CpG islands of LIN28A promoter in pancreatic cancer cells (Xu et al, 2016). In this study, we concentrated on the clinical relevance of MBD3 expression and its roles in the growth and EMT of pancreatic cancer cells. MBD3 expression is described for the first time in pancreatic cancer tissues in different clinicopathological features, at different tumour stages, and is associated with the outcome of patients. Previous studies have determined that MBD3 expression is downregulated in gastric carcinogenesis (Pontes et al, 2014), colon, and lung cancer (Zhu et al, 2004), indicating that reduced MBD3 expression is a potential marker of the carcinogenesis process. In this study, MBD3 expression is lowest in pancreas adenocarcinoma ductal, and reduces progressively with the increase of clinical stage in pancreatic cancer. It is indicated that the reduced MBD3 expression may have an important role in the development of pancreatic cancer and may be a biomarker for the detection of cancer development and progression, and directly associated with clinicopathological features. In addition, patients with lower MBD3 levels survive significantly less than those with higher levels, suggesting that downregulation of MBD3 expression is associated with the poor outcome of patients. It is implied that MBD3 may be a suppressor in pancreatic cancer progression.
It is well-known that EMT has an important role in pancreatic cancer progression (Wang et al, 2013). In this study, we identify that MBD3 knockdown upregulates mesenchymal markers and downregulates the epithelial marker expression of E-cadherin. Our findings confirm that MBD3 has a significant role in inhibiting EMT in pancreatic cancer cells. In addition, during the EMT process, cancer cells not only gain enhanced the ability of migration and invasion but also acquire cancer stem cell traits (Scheel and Weinberg, 2012; Du et al, 2016). Previous reports have shown that MBD3 knockout results in increasing the stemness of cells (Rais et al, 2013). Therefore, we speculate that inhibition of MBD3 on EMT might be partially via regulating stemness of cancer cells.
Previous study has demonstrated that TGF-β/Smad signalling is operational in pancreatic cancer (Togashi et al, 2014). When TGF-β is activated, Smad2 is phosphorylated and undergoes dimerisation with Smad3, thus allowing its translocation into nucleus in cancer cells, promoting the EMT progress (Liu et al, 2012; Liu et al, 2015). In this study, we find that MBD3 knockdown induces upregulating the TGF-β, p-Smad2/3; MBD3 overexpression results in downregulating the TGF-β, p-Smad2/3 in pancreatic cancer cells. Previously, MBD3 is a component of the Mi-2 autoantigen (Mi-2)/nucleosome remodelling and histone deacetylase (NuRD)-HDAC complex (Mi-2/nuclesome remodelling and deacetylase; Crook et al, 2006). MBD3 acts as a transcriptional repressor by recruiting HDACs, and is not only released from cancer-selective promoter of the histone deacetylase inhibitors (HDI) upregulated p21WAF1/Cip1 gene but also recruited to that of the HDI-downregulated ErbB2 gene, resulting in inhibiting the growth and survival of cancer cells (Noh et al, 2005). In addition, Snail is an important effector for the ability of invasion, forming Snail/HDAC1/HDAC2 repressor complex, and acting through transcriptional repression of E-cadherin during the EMT progress (von Burstin et al, 2009). Thus, it is reasonable to believe that Snail/HDAC1/HDAC2 repressor complex might be involved in inhibition of MBD3 on EMT. Furthermore, it is reported that HDAC1 is required for TGF-β-induced EMT (Lei et al, 2010), which might establish a link between MBD3 and TGF-β signalling. It is implied that MBD3 as a HDAC1 inhibiter suppress EMT probably via TGF-β/Smad signalling. Although, whether that the above two pathways are separated or associated remains unidentified, it is confirmed that MBD3 impair TGF-β/Smad signalling, and suppress EMT in pancreatic cancer cells. Importantly, HDI have been reported to be used in clinic, silencing of MBD3 abrogates the HDI-induced gene regulation and growth inhibition in cancer cells but not in normal cells (Noh et al, 2005). It is suggested that MBD3 has an important role in treating cancer and may be a potential therapeutic target candidate to pancreatic cancer.
In summary, our study provides proof that MBD3 inhibits EMT in pancreatic cancer cells probably through downregulating TGF-β/Smad signalling. All these findings suggest that MBD3 is a potential biomarker for diagnostics and prognosis of pancreas colloid type carcinoma and pancreatic cancer at stage 3 and stage 4.
Change history
03 January 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amundadottir LT (2016) Pancreatic Cancer Genetics. Int J Biol Sci 12 (3): 314–325.
Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I (2008) Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 55 (88): 2016–2027.
Brumbaugh J, Hochedlinger K (2013) Removing reprogramming roadblocks: Mbd3 depletion allows deterministic iPSC generation. Cell Stem Cell 13 (4): 379–381.
Cai G, Wu D, Wang Z, Xu Z, Wong KB, Ng CF, Chan FL, Yu S (2016) Collapsin response mediator protein-1 (CRMP1) acts as an invasion and metastasis suppressor of prostate cancer via its suppression of epithelial-mesenchymal transition and remodeling of actin cytoskeleton organization. Oncogene e-pub ahead of print 20 June 2016 doi:10.1038/onc.2016.227.
Choi WI, Jeon BN, Yoon JH, Koh DI, Kim MH, Yu MY, Lee KM, Kim Y, Kim K, Hur SS, Lee CE, Kim KS, Hur MW (2013) The proto-oncoprotein FBI-1 interacts with MBD3 to recruit the Mi-2/NuRD-HDAC complex and BCoR and to silence p21WAF/CDKN1A by DNA methylation. Nucleic Acids Res 41 (13): 6403–6420.
Crook JM, Dunn NR, Colman A (2006) Repressed by a NuRD. Nat Cell Biol 8 (3): 212–214.
Del Chiaro M, Segersvard R, Lohr M, Verbeke C (2014) Early detection and prevention of pancreatic cancer: is it really possible today? World J Gastroenterol 20 (34): 12118–12131.
Du L, Yamamoto S, Burnette BL, Huang D, Gao K, Jamshidi N, Kuo MD (2016) Transcriptome profiling reveals novel gene expression signatures and regulating transcription factors of TGFbeta-induced epithelial-to-mesenchymal transition. Cancer Med 5 (8): 1962–1972.
Elsasser HP, Lehr U, Agricola B, Kern HF (1992) Establishment and characterisation of two cell lines with different grade of differentiation derived from one primary human pancreatic adenocarcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol 61 (5): 295–306.
Gunasinghe NP, Wells A, Thompson EW, Hugo HJ (2012) Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev 31 (3-4): 469–478.
Karaczyn AA, Adams TL, Cheng RY, Matluk NN, Verdi JM (2016) Human NUMB6 induces epithelial-mesenchymal transition and enhances breast cancer cells migration and invasion. J Cell Biochem e-pub ahead of print 15 June 2016 doi:10.1002/jcb.25628.
Klein AP (2012) Genetic susceptibility to pancreatic cancer. Mol Carcinog 51 (1): 14–24.
Kyriazis AP, McCombs WB 3rd, Sandberg AA, Kyriazis AA, Sloane NH, Lepera R (1983) Establishment and characterization of human pancreatic adenocarcinoma cell line SW-1990 in tissue culture and the nude mouse. Cancer Res 43 (9): 4393–4401.
Lei W, Zhang K, Pan X, Hu Y, Wang D, Yuan X, Shu G, Song J (2010) Histone deacetylase 1 is required for transforming growth factor-beta1-induced epithelial-mesenchymal transition. Int J Biochem Cell Biol 42 (9): 1489–1497.
Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M, Todaro G (1975) Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int J Cancer 15 (5): 741–747.
Liu LC, Tsao TC, Hsu SR, Wang HC, Tsai TC, Kao JY, Way TD (2012) EGCG inhibits transforming growth factor-beta-mediated epithelial-to-mesenchymal transition via the inhibition of Smad2 and Erk1/2 signaling pathways in nonsmall cell lung cancer cells. J Agric Food Chem 60 (39): 9863–9873.
Liu XN, Wang S, Yang Q, Wang YJ, Chen DX, Zhu XX (2015) ESC reverses epithelial mesenchymal transition induced by transforming growth factor-beta via inhibition of Smad signal pathway in HepG2 liver cancer cells. Cancer Cell Int 15: 114.
Menafra R, Stunnenberg HG (2014) MBD2 and MBD3: elusive functions and mechanisms. Front Genet 5: 428.
Noh EJ, Jang ER, Jeong G, Lee YM, Min CK, Lee JS (2005) Methyl CpG-binding domain protein 3 mediates cancer-selective cytotoxicity by histone deacetylase inhibitors via differential transcriptional reprogramming in lung cancer cells. Cancer Res 65 (24): 11400–11410.
Pontes TB, Chen ES, Gigek CO, Calcagno DQ, Wisnieski F, Leal MF, Demachki S, Assumpcao PP, Artigiani R, Lourenco LG, Burbano RR, Arruda Cardoso Smith M (2014) Reduced mRNA expression levels of MBD2 and MBD3 in gastric carcinogenesis. Tumour Biol 35 (4): 3447–3453.
Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, Maza I, Mor N, Baran D, Weinberger L, Jaitin DA, Lara-Astiaso D, Blecher-Gonen R, Shipony Z, Mukamel Z, Hagai T, Gilad S, Amann-Zalcenstein D, Tanay A, Amit I, Novershtern N, Hanna JH (2013) Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502 (7469): 65–70.
Scheel C, Weinberg RA (2012) Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22 (5-6): 396–403.
Tam WL, Weinberg RA (2013) The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 19 (11): 1438–1449.
Thota R, Pauff JM, Berlin JD (2014) Treatment of metastatic pancreatic adenocarcinoma: a review. Oncology 28 (1): 70–74.
Togashi Y, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, Fujita Y, Kodera Y, Sakai K, Tomida S, Kitano M, Ito A, Kudo M, Nishio K (2014) Homozygous deletion of the activin A receptor, type IB gene is associated with an aggressive cancer phenotype in pancreatic cancer. Mol Cancer 13: 126.
von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, von Werder A, Schmidt A, Mages J, Pagel P, Schnieke A, Schmid RM, Schneider G, Saur D (2009) E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 137 (1): 361–371, 371 e1-5.
Wang Z, Ali S, Banerjee S, Bao B, Li Y, Azmi AS, Korc M, Sarkar FH (2013) Activated K-Ras and INK4a/Arf deficiency promote aggressiveness of pancreatic cancer by induction of EMT consistent with cancer stem cell phenotype. J Cell Physiol 228 (3): 556–562.
Wood KH, Zhou Z (2016) Emerging molecular and biological functions of MBD2, a reader of DNA methylation. Front Genet 7: 93.
Xu M, Bian S, Li J, He J, Chen H, Ge L, Jiao Z, Zhang Y, Peng W, Du F, Mo Y, Gong A (2016) MeCP2 suppresses LIN28A expression via binding to its methylated-CpG islands in pancreatic cancer cells. Oncotarget 7 (12): 14476–14485.
Yokoyama S, Higashi M, Kitamoto S, Oeldorf M, Knippschild U, Kornmann M, Maemura K, Kurahara H, Wiest E, Hamada T, Kitazono I, Goto Y, Tasaki T, Hiraki T, Hatanaka K, Mataki Y, Taguchi H, Hashimoto S, Batra SK, Tanimoto A, Yonezawa S, Hollingsworth MA (2016) Aberrant methylation of MUC1 and MUC4 promoters are potential prognostic biomarkers for pancreatic ductal adenocarcinomas. Oncotarget 7 (27): 42553–42565.
Yoshida K, Murata M, Yamaguchi T, Matsuzaki K, Okazaki K (2016) Reversible human TGF-beta signal shifting between tumor suppression and fibro-carcinogenesis: implications of Smad phospho-isoforms for hepatic epithelial-mesenchymal transitions. J Clin Med 5 (1).
Zagorac S, Alcala S, Fernandez Bayon G, Bou Kheir T, Schoenhals M, Gonzalez-Neira A, Fernandez Fraga M, Aicher A, Sainz B, Heeschen C (2016) DNMT1 inhibition reprograms pancreatic cancer cells via upregulation of the miR-17-92 cluster. Cancer Res 76 (15): 4546–4558.
Zhang L, Zheng Y, Sun Y, Zhang Y, Yan J, Chen Z, Jiang H (2016) MiR-134-Mbd3 axis regulates the induction of pluripotency. J Cell Mol Med 20 (6): 1150–1158.
Zhu Y, Harrison DJ, Bader SA (2004) Genetic and epigenetic analyses of MBD3 in colon and lung cancer. Br J Cancer 90 (10): 1972–1975.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81672402, 81472333 and 81372718) and the Natural Science Foundation of Jiangsu Province (BK20131247).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Xu, M., He, J., Li, J. et al. Methyl-CpG-binding domain 3 inhibits epithelial–mesenchymal transition in pancreatic cancer cells via TGF-β/Smad signalling. Br J Cancer 116, 91–99 (2017). https://doi.org/10.1038/bjc.2016.397
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.397
Keywords
This article is cited by
-
MBD3 promotes epithelial-mesenchymal transition in gastric cancer cells by upregulating ACTG1 via the PI3K/AKT pathway
Biological Procedures Online (2024)
-
MBD3 promotes hepatocellular carcinoma progression and metastasis through negative regulation of tumour suppressor TFPI2
British Journal of Cancer (2022)
-
PBX3/MEK/ERK1/2/LIN28/let-7b positive feedback loop enhances mesenchymal phenotype to promote glioblastoma migration and invasion
Journal of Experimental & Clinical Cancer Research (2018)
-
SHCBP1 promotes synovial sarcoma cell metastasis via targeting TGF-β1/Smad signaling pathway and is associated with poor prognosis
Journal of Experimental & Clinical Cancer Research (2017)
