
Abstract
Background:
The Hedgehog (Hh) pathway is upregulated in cervical cancer and associated with poor outcome. We explored the effects of Hh pathway inhibition in combination with RTCT in a patient derived orthotopic cervical cancer xenograft model (OCICx).
Methods:
5E1, a monoclonal antibody for SHH, or Sonidegib (LDE225), a clinical SMO inhibitor (Novartis) were added to RTCT. We investigated tumour growth delay, metastasis and GI toxicity using orthotopic cervical cancer xenografts models. The xenografts were treated with radiotherapy (15 × 2 Gy daily fractions over 3 weeks) and weekly cisplatin 4 mg kg−1 concurrently, with or without 5E1 or Sonidegib (LDE225). The Hh inhibitors were administered by subcutaneous injection (5E1; 20 mg kg−1 weekly for 3 weeks), or by oral gavage (Sonidegib; 60 mg kg−1 daily for 3 weeks).
Results:
We observed that both Hh inhibitors administered with RTCT were well tolerated and showed increased tumour growth delay, and reduced metastasis, with no increase in acute GI-toxicity relative to RTCT alone.
Conclusions:
Our data suggest Hh can be a valid therapeutic target in cervical cancer and supports data suggesting a potential therapeutic role for targeting Hh in patients undergoing RTCT. This warrants further investigation in clinical trials.
Similar content being viewed by others


Main
Worldwide, cervical carcinoma is the fourth leading cause of cancer-related death in women (Jemal et al, 2011). The incidence of cervical cancer has declined in the developed world following the introduction of screening programs, a decline which is expected to continue with the increased availability of HPV vaccination. Globally, however, it remains a major cause of cancer-related morbidity and mortality (Statistics CC, 2015). Within the United States the proportion of women presenting with later stage disease is increasing, disproportionately affecting medically under-resourced sections of society (Funke and Silberstein, 2015). There remains, therefore, an ongoing need to optimise the frontline treatment of this disease. Standard of care for the primary treatment of women diagnosed with stage IB-IIIB cervical cancer consists of a combination of platinum-based chemotherapy delivered concurrently with radiation (RTCT) and has, from a systemic therapy perspective, remained largely unchanged since the 1990s (Greer et al, 2010). Current RTCT regimens are being delivered close to (or at) the limits of normal tissue tolerance. Thus, further dose intensification of chemotherapy or the addition of further cytotoxic agents in combination with radiation is not likely to be a successful strategy. There is therefore an urgent unmet need to explore the potential of combining, rationally chosen, targeted agents with RTCT for the treatment of cervical cancer.
Conducting early phase clinical trials in a potentially curable patient population poses significant ethical and regulatory challenges. Furthermore, given the wide range of potential targets and agents, it is essential that in a tumour type such as cervical cancer where there is a limited pool of potential patients, any trials pursued need to be underpinned by high-quality pre-clinical data in a relevant model. Identification of potentially druggable targets and the acquisition of pre-clinical efficacy data are essential. If we are to move into early phase frontline clinical trials, exploration of fractionated radiotherapy schedules and indicators of potential toxicity are also of key importance (Harrington et al, 2011). The use of early passage orthotopic patient-derived cervical cancer xenograft models as developed in our Institute (OCICx) potentially allows us to achieve this, as these models recapitulate not only the metastatic behaviour of cervix cancers but also retain the tumour heterogeneity and stromal characteristics of the original tumour (Chaudary et al, 2012, 2015). They afford the opportunity of studying fractionated radiotherapy and multi-dose chemotherapy regimens delivered in a manner similar to the treatment of patients in the clinic (Chaudary et al, 2011).
The Hedgehog (Hh) signalling pathway controls cell proliferation and differentiation during embryonic development, but is largely suppressed in the adult. Emerging data support critical roles for Hh activation in carcinogenesis, promoting the cancer stem cell phenotype, epithelial to mesenchymal transition (EMT) and metastasis. Aberrant activation of the pathway occurs in several disparate tumours, including cervical cancer (Ingham and McMahon, 2001; Chaudary et al, 2012b; Justilien and Fields, 2015). The Hh pathway has been implicated in DNA repair and its activation has been proposed as a mechanism for resistance to both chemotherapy and radiation (Meng et al, 2015). Hh pathway activation is initiated by binding of one of the three Hh ligand proteins: Sonic hedgehog (SHH); Indian Hedgehog (IHH) or Desert Hedgehog, to Patched (PTCH1/2). In the absence of Hh ligand PTCH functions as a tumour suppressor inhibiting Smoothened (SMO). Binding of any of the three Hh ligands to PTCH relieves the suppression of SMO resulting in dissociation of a cytoplasmic inhibitory complex that targets the glioma-associated oncogene homologue GLI family of transcription factors for proteolytic cleavage (Ingham and McMahon, 2001; Hooper and Scott, 2005). Translocation of GLI to the nucleus affects a number of cellular functions including promotion of cell cycle progression, EMT, anti-apoptotic proteins and production of vascular endothelial growth factor (VEGF) (Pasca di Magliano and Hebrok, 2003; Hooper and Scott, 2005; Shevde and Samant, 2014; Azzi et al, 2015). Studies in a number of tumour types indicate stromal activation of Hh may be critical in tumourigenesis (Yauch et al, 2008; Meerang et al, 2016).
Several models for Hh pathway activation in cancer have been proposed: ligand dependent, autocrine; ligand dependent, paracrine (stromal tumour interaction) or ligand independent, mutation driven (Gorlin’s Syndrome; Scales and de Sauvage, 2009) Hh pathway activation in cancer can occur in tumour cells and/or in the stroma depending on the model and this appears to differ between tumour types (Scales and de Sauvage, 2009).
We have previously shown, using real-time quantitative PCR (RT-PCR), that upregulation of one or more members of the Hh pathway occurs in cervical cancers compared to normal cervix tissue. We demonstrated that upregulation of SMO and of >3 of the Hh pathway members was associated with a worse recurrence-free patient survival (Chaudary et al, 2012b). Bohr Mordhorst et al (2014) also demonstrated, using immunohistochemistry, an association between poor outcome and expression of PTCH, SMO and GLi2. The aim of the current study, using early passage orthotopic, patient-derived, cervical cancer xenograft models, was to further define the role of the Hh pathway in cervical cancer and to investigate the therapeutic potential of Hh inhibition in combination with fractionated radiation and chemotherapy.
Materials and methods
Orthotopic xenograft models of cervical cancer
Development, engraftment and stromal characteristics of our patient-derived, cervical cancer xenograft models (OCICx) have been previously described (Chaudary et al, 2011, 2012, 2015). The OCICx models were developed from cervical cancer samples taken from patients (prior to definitive therapy) at Princess Margaret Cancer Centre, participating in a prospective translational research program. Patients signed consent according to a protocol approved by the University Health Network/University of Toronto Research Ethics Board. The OCICx models were grown in the cervix of NOD-SCID or (for irradiation studies) in NOD-Rag1nullIL2rγnull (NRG) immune-deprived female mice 6–8 weeks old. ME180 cells, a human cervical carcinoma cell line, were cultured in 10% fetal bovine serum in alpha MEM and donor pieces (2–3 mm2- generated from intramuscular tumours grown in the hind leg of a mouse) were sutured directly onto the cervix as described previously (Cairns and Hill, 2004; Chaudary et al, 2011, 2012; Justilien and Fields, 2015). All animal experiments were performed in accordance with institutional Animal Care Committee guidelines.
Hh gene expression in OCICx models
We have previously reported Hh gene expression in cervical cancer samples from 96 patients compared to normal tissue using qRT-PCR (10). Similarly, we used this technique to examine Hh gene expression in the OCICx models, examining both murine and human IHH, SHH, SMO, PTCH1, PTCH2, GLi1 gene expression levels. Total RNA was extracted from frozen tissue sections using the Qiagen RNeasy Mini Extraction kit (Qiagen, Mississauga) and real-time PCR was performed according to the manufacturer’s instructions as previously described (Chaudary et al, 2012b). Human L32, ribosomal protein, was used as an endogenous control for normalisation. Samples were run in triplicate for each sample and normalised against L32. All analyses were performed blinded to study end point. For gene expression studies following drug treatment ME180 tumours were treated with 5E1 for 21 days before analysis or treated with LDE225 for 7 days before analysis. In both cases treatment started at Day 9 after implant (4–5 mm).
Radiation and tumour growth delay studies
Growth delay experiments were performed on orthotopic ME180 tumours and 3 OCICx models 29, 30 and 34 (at passage 3) selected to recapitulate the different Hh expressions patterns we observed in our series of patients and to reflect tumours with differing stromal content (range of 10% in ME180 to >60% in OCICx 29), as shown in Figure 1. The clinical status of the three cervical cancer patients, from which these xenografts models were developed, are described in Supplementary Table 1. All tumours were of squamous cell carcinoma histology and were associated with either HPV 16 or 18 infection.

(A) Hh gene expression in cervical cancer xenografts. The qRT-PCR data sets (mean±s.e.) for OCICx models 29, 30, 34 and ME180 are shown for human and mouse Hh genes: IHH, SHH, SMO, PTCH1, PTCH2, GLi1. Data sets were normalised against the L32 housekeeping gene. (B) The per cent stroma levels as defined by Spectrum Genie Aperio analysis (Ludwig and Weinstein, 2005) with H&E representative images for the 3 OCICx and ME180 tumours.
Mice were imaged and irradiated using a small animal irradiator with integrated cone beam computed tomography (CBCT; X-Rad 225Cx, Precision X-ray, North Branford, CT, USA) described previously (Clarkson et al, 2011). Following implantation in the cervix, biweekly tumour measurements were made using CBCT. The mice were randomly assigned to one of five groups when the tumours reached a diameter of 4–5 mm: control (no treatment), radiotherapy alone (15 × 2 Gy daily fractions over 3 weeks), RTCT (same radiotherapy+weekly cisplatin 4 mg kg−1 intraperitoneally given within 1 h before the radiation fraction at the beginning of each week), RTCT plus Hh inhibitor (SHH IgG antibody 5E1; 20 mg kg−1 subcutaneously weekly for 3 weeks before each radiation treatment; OICR Cancer Stem Cell Program); or SMO inhibitor (LDE225 (Sonidegib) 60 mg kg−1 by oral gavage daily for 3 weeks; Novartis; Figure 2). For radiation treatment, mice were positioned in a specialised jig, imaged and adjusted to assure reproducible tumour targeting. An optimised treatment plan was used with eight circular beams equally positioned around the tumour (Supplementary Figure 1). The X-ray tube was calibrated at 225 kVp, 13 mA (HVL: 0.93 mm Cu, added filtration: 0.3 mm Cu) following the TG-61 protocol (Ma et al, 2001; van Hoof et al, 2013). The radiation dose rate was ∼3.0 Gy min−1. The dose protocol for the SMO inhibitor was chosen based on previously published reports in vivo (Steg et al, 2012; O'Reilly et al, 2013). The treatment protocol is summarised schematically in Figure 2. Tumour size and local progression after treatment were assessed weekly using the CT imaging capability of the irradiator. Mice were killed at a primary tumour size of 1–1.5 cm. Animal weights were monitored daily during treatment and weekly after treatment until the end of the experiment (tumour regrowth). Metastatic progression to para-aortic nodes was assessed at the time that the mice were killed based on lymph node size and H&E histology.

The experimental design is shown in the schematic, indicating the time of tumour implants, treatment window and the monitoring of tumour regrowth.
Acute gastrointestinal toxicity
To evaluate acute gastrointestinal toxicity, a frequent side effect in patients receiving RTCT for cervical cancer, a gut colony assay was used according to the method described by Withers and Elkind (Tucker et al, 1983; Withers and Elkind, 1970). Cisplatin and/or Hh inhibitor, at the doses given above, were given immediately before a single radiation dose of 10, 11, 12 or 14 Gy. Whole body radiation was administered with an XRAD 320 Precision Xray machine to non-tumour-bearing C57BL/6 female mice and the animals were sacrificed 3.5 days post-treatment. The jejunum was removed, flushed with PBS, fixed in formalin and stained for Ki67 (proliferation marker) to identify crypts that contained proliferating cells (n>5 Ki67 positive cells) using Aperio Image Scope (Gani et al, 2015).
Statistical analysis
The statistical significance of differences between experimental groups in the lymph node metastasis data and 5E1 and LDE225 Hh gene expression levels relative to control were analysed using one-way ANOVA with Newman-Keuls Multiple Comparison test (Graph-Pad 5 Software). Results with a P-value<0.05 were considered statistically different. For the growth delay experiments, the time for the tumour to double relative to the size at initiation of radiation treatment was determined. These time points were compared between treatments using a nonparametric test: Kruskal-Wallis for the comparisons between all five treatments and Mann-Whitney for the comparisons between the groups.
Results
Hh gene expression in cervical cancer xenografts reflects expression seen in patient samples
Varying levels of human Hh ligand and receptor expression, IHH, SHH, SMO, PTCH1, PTCH2, GLi1, were observed in the OCICx models as they had been in the primary tumours. Murine Hh gene expression was measured in addition to human Hh gene expression (Figure 1A) and was found to be more marked in the OCICX models compared to the ME180 orthotopic xenograft model, which has low stromal content (Figure 1B). The murine Hh gene expression levels reflected the higher stromal content of the OCICx models as assessed by histology. We selected 3 OCICx models (29, 30 and 34), which varied in stromal content in addition to the cervical cell line ME180 for further study (Figure 1B). We observed that treatment with 5E1 and LDE225 (independently) reduced human Hh gene expression in ME180 tumours (Figure 3).

Hh gene expression in ME180 orthotopic cervix tumours treated with Hh inhibitors: 5E1 and LDE225. 5E1 was administered weekly for 21 days in treatment (20 mg kg i.p subcutaneously). The LDE225 was administered daily for 7 days (60 mg kg by oral gavage). The qRT-PCR data set (mean±s.e. with 4–5 mice per group) for human IHH, SHH, SMO, PTCH1, PTCH2, GLi1 is shown. Data sets were normalised against the L32 housekeeping gene. Treatment was started at Day 9 post implant at tumour size of 4–5 mm. Hh expression levels with 5E1 and LDE225 treatment show significance relative to control. P<0.05.
SHH inhibition via 5E1 antibody in combination with RTCT resulted in tumour growth delay and reduced metastasis in OCICx models
Tumour growth delay studies with the SHH inhibitor, 5E1, were conducted in ME180, OCICx 30, and OCICx 34 models (Figure 4; Supplementary Table 2). There was no benefit of 5E1 alone compared to untreated (control) tumours in any of the tumour models. The results with the ME180 cell line xenografts did not demonstrate any benefit from the addition of 5E1 to RTCT (P=0.8). However, in the OCICx models, the addition of 5E1 to RTCT resulted in greater tumour growth delay (P=0.029 for both) and prolonged survival compared with RTCT alone. The combination of 5E1 with RTCT also resulted in a reduction in the metastatic burden in para-aortic lymph nodes (Figure 4B) compared with RTCT alone in both the OCICx 30 (P<0.01) and OCICx 34 (P<0.001) models. The ME 180 tumour showed a similar trend.

5E1 enhanced the efficacy of cisplatin-based chemoradiotherapy in OCICx xenograft tumours 30 and 34 and ME180 tumours. (A) Tumour growth curves after treatment are shown with fractionated X-ray irradiation (RT- 2Gy daily × 15 fractions; 30 Gy) and cisplatin 4 mg kg weekly for 3 weeks, RT alone, 5E1 alone (20 mg.kg i.p subcutaneously weekly for 3 weeks) or in combination (RT+CT+5E1). When tumours reached the 3–4 mm in size, the mice were randomised into 4–5 mice per group. Individual growth curves are shown for each mouse in all groups. Dotted line represents the end of the treatment window. Tumour volume relative to initial treatment volume (5 mice per group); dotted vertical line indicates treatment window up to Day 21. (B) Mean number of positive nodes per mouse in each group is plotted (mean±s.e.). (C) Survival plots are shown for each model in each treatment group. The animals were killed when the tumours reached a size of 1.5 cm largest diameter.
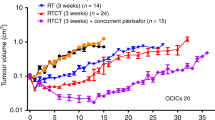
SMO inhibition via LDE225 (Sonidegib) in combination with RTCT resulted in tumour growth delay and reduced metastasis
Tumour growth delay studies with the SMO inhibitor, LDE225 (Sonidegib), were conducted in OCICx 29, and OCICx 34 models (Figure 5). The control and LDE225 (drug alone) showed no difference in tumour growth. The addition of LDE225 to RTCT resulted in significant tumour growth delay (P=0.029) and improved animal survival in OCICx 29 but, although there was a trend for a similar effect in OCICx 34, it was not statistical significant (Supplementary Table 2). The tumours decreased in volume post the 3 week treatment however the regrowth time varied for the two models. The OCICx 34 xenografts initially regrew more rapidly compared to the OCICx 29 xenografts in the RTCT+LDE225 treatment arm, although their growth slowed as they became larger. A significantly reduced metastatic burden in para-aortic lymph nodes was also observed with the addition of LDE225 to RTCT in model OCICx 29 (P<0.001) but not in OCICx 34 (Figure 5).

LDE225 enhanced the efficacy of cisplatin-based chemoradiotherapy in OCICx xenograft tumours 29 and 34. (A) Tumour growth curves after treatment are shown with fractionated X-ray irradiation (RT- 2Gy daily × 15 fractions; 30 Gy) and cisplatin 4 mg kg weekly for 3 weeks, RT alone, LDE225 alone (LDE225; 60 mg kg o.g; × 3 weeks) or in combination (RT+CT+LDE225). When tumours reached the 3–4 mm in size, the mice were randomised into 4–5 mice per group. Individual growth curves are shown for each mice in all groups. Dotted line represents the end of the treatment window up to Day 21. Tumour volume relative to initial treatment volume; 5 mice per group; dotted line indicates end of treatment. (B) Mean number of positive nodes/mouse in each group is plotted (mean±s.e.). (C) Survival plots are shown for each model in each treatment group. The animals were killed when the tumours reached a size of 1.5 cm largest diameter.
Inhibition of the Hedgehog pathway in combination with fractionated radiation and concurrent cisplatin does not result in increased acute gastrointestinal toxicity
The addition of either 5E1 or LDE225 to RTCT did not cause increased weight loss post treatment compared with RTCT alone. There was a slight reduction in weight during treatment but the weights recovered well afterwards in both models (data not shown). A pathologist (M Larson, Mbed Pathology) examined normal organs adjacent to the irradiated volume (rectum, small bowel, bladder) for toxicity. There was no gross or microscopic (H&E sections) evidence of excessive injury with the addition of either 5E1 or LDE225 to RTCT alone.
The potential toxicity of LDE225 or 5E1 and the combination of drug with RTCT was also assessed using an acute gut colony assay(Tucker et al, 1983; Withers and Elkind, 1970). The effects of treatment on gut toxicity were evaluated using single dose irradiation (10–14 Gy) to the whole mouse with concurrent cisplatin (4 mg kg−1) combined with either 5E1 or LDE225. The number of regenerating crypts in the jejunum was assessed 3.5 days after treatment to determine survival curves. As shown in Figure 6, neither 5E1 nor LDE225 administered alone caused detectable loss of proliferating crypts. The combination of RT and cisplatin produced significantly greater crypt loss than RT alone. The addition of either 5E1 or LDE225 to RTCT and or RT alone appeared to have a protective effect on crypt cell survival (P<0.001 for both drugs) after a large single dose of 14 Gy.

Acute gut toxicity with Hh treatment. Gut colony assay for acute toxicity with Cisplatin and/or Hh (5E1 or LDE225) was given immediately prior to radiation at doses of 10, 11, 12, 14 Gy. Whole body radiation was administered in non–tumour bearing NRG female mice and the animals were sacrificed 3.5 days post treatment. The mean values plotted (6 gut crypts per mouse; n=4–5 mice per treatment group) are per cent survival of Ki-67 (proliferation marker), by immunohistochemistry, positive gut crypts that contain proliferating cells. * indicates significance of P<0.05 relative to Control within each radiation dose, ** indicates significance relative to rad+cis.^ indicates significance between the 14 Gy radiation dose compared to 10–12 Gy doses.
Discussion
We have previously demonstrated Hh pathway upregulation in tumours taken from patients with cervical cancer compared with normal cervical tissue (Chaudary et al, 2012b). Our data, and that of others, have shown that increased expression of Hh pathway members before definitive RTCT is associated with poor outcome (Chaudary et al, 2012b; Bohr Mordhorst et al, 2014). Here we explored the Hh pathway in the context of a unique orthotopic, patient-derived cervical cancer pre-clinical model that exhibits the stromal characteristics and metastatic behaviour of cervical cancer in the clinic (Chaudary et al, 2011, 2012b). The OCICx models recapitulated the increased expression of the Hh pathway seen in the patient samples and were chosen to reflect a range of stromal content and levels of HH expression. Both mouse and human Hh gene expression in the OCICx models was observed. This suggests the possibility of a ligand dependent, paracrine model of Hh pathway activation in cervical cancer as in the OCICx models stroma tends to be murine rather than of human origin unlike the tumour cells. In the ME180 cell line orthotopic xenograft model, increased Hh pathway expression was predominantly human in keeping with the very low stromal content (Figure 1). Using our OCICx models we were able to deliver fractionated radiotherapy and weekly doses of cisplatin (RTCT) in a schedule that mimics treatment delivered in the clinic. When the Hh inhibitors, 5E1 or LDE225, were combined with RTCT there was delayed tumour growth, prolonged survival of the animals and a reduction in lymph node metastases at time of killing. Furthermore, no additive acute toxicity was observed for the combination of Hedgehog inhibition with RTCT suggesting that this may be well tolerated in patients.
Most xenograft models of cervical cancer have been developed using commercially available cell lines that have limitations, notably poor correlation between the biological characteristics of a tumour in a patient and the corresponding cell line because of genetic instability and multiple passaging (Whatcott et al, 2015). Commercially available cell lines when grown in mice may not adequately represent clinical characteristics or the behaviour of human tumours (Hidalgo et al, 2014; Boone et al, 2015). The establishment of patient-derived cervical cancer xenograft models utilising samples taken from patients before treatment potentially addresses some of these concerns. Most xenograft models are generated by subcutaneously implantation, as the accessibility of this site contributes to the relative ease of developing and testing novel agents. However, in these models the microenvironment of subcutaneous, or intraperitoneal, murine models may not reflect that of the original tumour (John et al, 2011). Recapitulation of the original tumour microenvironment has a greater likelihood of occurring in orthotopic models. We have previously demonstrated that our OCICx models show a relatively stable retention of the original tumour characteristics including stromal content and tumour heterogeneity (Chaudary et al, 2011, 2012b, 2015).
Recapitulation of the tumour microenvironment is of particular importance when investigating potentially druggable pathways that affect the tumour microenvironment, such as the Hedgehog inhibitors. This can be seen when comparing gene expression, stromal content and ultimately the response to treatment between OCICx models and the ME180 cell line xenografts. Stromal content is low in ME180 and is not representative of that seen in patient samples or in some of the primary xenografts. OCICx 34, which has the lowest stromal content (20%) of the patient derived models, showed limited response after RTCT+LDE225 and there was no reduction in lymph node metastases compared with RTCT alone, in contrast to OCIC 29 with higher stromal content. These differences in tumour response to Hh inhibition may link directly to differences in stromal content or potentially differential expression of the Hh pathway components. Stromal content combined with Hh expression within the tumour could be explored as a predictor of response in the clinical setting. One limitation of this model, however, is that the mice used are, of necessity, immune incompetent. Data suggest that the Hh pathway may play a role in regulating T cell response (Hanna and Shevde, 2016) and this effect would not be captured by our model.
Inhibition of Hh has been a successful therapeutic strategy in basal cell carcinoma, which exhibits ligand independent activation of the Hh pathway secondary to an activating mutation (Basset-Seguin et al, 2015). In other tumours, which do not obviously exhibit ligand-independent activation, targeting the Hh pathway has met with less success. Initially, targeting the Hh pathway was felt to be a promising option for the treatment of pancreatic cancer where upregulation of SHH has been reported in over 70% of tumours. In pancreatic cancer, as in cervical cancer, a ligand dependent paracrine mechanism appears to be responsible for activation of the Hh pathway (Onishi and Katano, 2014). In 2008, Olive et al (2009) studied the effect of IPI-926 (a SMO inhibitor) in a pancreatic cancer pre-clinical model. They provided proof-of-principle that inhibition of the Hh pathway could disrupt the desmoplastic stroma, facilitating the delivery and enhancing the efficacy of chemotherapy. This led to a number of clinical trials in this disease. While early indications of efficacy from phase I studies were promising, data from the phase II study (NCT01130142) suggested a worse outcome for patients treated with the combination of Hh inhibitor and chemotherapy (Ko et al, 2016). Rhim et al (2009) explored this paradox in a mouse model showing that chronic depletion of Hh activity either as a result of deletion of SHH or prolonged administration of a Hh inhibitor resulted in accelerated tumour growth, more macrometastatic disease and shorter survival times. Clearly in pancreas cancer the dose and scheduling in the pre-clinical models was critical for understanding the potential outcome in the clinical study. A number of theories have been proposed to explain this effect. Chronic depletion of Hh, may lead to remodelling of the stroma to such an extent that it removes a constraint to tumour growth; whilst a second theory proposes that the stage of the tumour and the context of the treatment may be critical to the efficacy of Hh inhibition (Olive et al, 2009; Rhim et al, 2009). Differences in stromal content between the primary tumour and metastatic deposits may impact the potential efficacy of Hh inhibition. The lower stromal content observed in some metastases compared to the primary tumour may limit the efficacy of Hh inhibition in advanced or metastatic disease (Whatcott et al, 2015). Considering these issues in the context of our pre-clinical cervical cancer experiments, we added short term (3weeks) Hh inhibition to standard RTCT for localised, treatment-naïve disease and demonstrated improvements in local tumour response and reduced metastases. This approach, focused on the treatment of earlier, potentially curable, disease may have a greater chance of success going forward into the clinic.
Emerging data on the relationship between DNA repair and the Hh pathway suggests that inhibition of the activity of GLI can interfere with almost all types of DNA repair in human cancer, indicating that Hh/GLI functions may play an important role in enabling tumour cells to survive types of DNA damage induced by RTCT (Meng et al, 2015). GLI1 also plays a pivotal role in cellular accumulation of cisplatin in cisplatin-resistant A2780-CP70 human ovarian cancer cells (Amable et al, 2014). Pretreatment of the cisplatin-resistant human ovarian cancer cell line A2780-CP70 with anti-GLI1 shRNA resulted in supra-additive cell killing with cisplatin. We have previously shown that precision irradiation in oesophageal PDX models increases Hh gene expression with PTCH1,2 and GLI1 upregulated in the stroma (Teichman, 2012). Our pre-clinical data supports the cell line and mechanistic studies in terms of the potential for additive benefit from combining Hh inhibition with RTCT.
The availability of image-guided small animal irradiator technology meant that the orthotopic, primary mouse xenograft models could be treated with fractionated radiation alone and in combination with weekly cisplatin chemotherapy in a manner that mimics clinical regimens (Chaudary et al, 2014). This allowed us to not only study the efficacy of combination but also to evaluate infield toxicity (both early and late effects). This is highly relevant especially if we are to consider early phase clinical trials in the frontline, curative setting (Withers and Mason, 1974; Withers et al, 1974). Our data suggest that the combination of RTCT with Hedgehog inhibition is likely to be well tolerated. Moving this into a clinical trial would require development of an early phase protocol involving a high risk population and incorporating high quality translational studies. The pre-clinical data presented here is limited to 4 models, however, they suggest that stromal content may be a potential predictive biomarker and investigation in an unselected trial population would allow further study of this possibility. Interactions with hypoxia and non-classical Hh pathway activation would also need to be considered.
In conclusion, this study, using the unique OCICx primary xenograft cervical cancer models, demonstrates that the combination of Hh inhibition with RTCT can induce tumour growth delay that persists weeks after the end of treatment and reduces lymph node metastases compared to RTCT alone. This is a potentially attractive strategy to develop in the clinic for the treatment of cervical cancer.
Change history
03 January 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amable L, Fain J, Gavin E, Reed E (2014) Gli1 contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncol Rep 32 (2): 469–474.
Azzi S, Treps L, Leclair HM, Ngo HM, Harford-Wright E, Gavard J (2015) Desert Hedgehog/Patch2 axis contributes to vascular permeability and angiogenesis in glioblastoma. Front Pharmacol 6: 281.
Basset-Seguin N, Hauschild A, Grob JJ, Kunstfeld R, Dreno B, Mortier L, Ascierto PA, Licitra L, Dutriaux C, Thomas L, Jouary T, Meyer N, Guillot B, Dummer R, Fife K, Ernst DS, Williams S, Fittipaldo A, Xynos I, Hansson J (2015) Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-planned interim analysis of an international, open-label trial. Lancet Oncol 16 (6): 729–736.
Bohr Mordhorst L, Ahlin C, Sorbe B (2014) Prognostic impact of the expression of Hedgehog proteins in cervical carcinoma FIGO stages I-IV treated with radiotherapy or chemoradiotherapy. Gynecol Oncol 135 (2): 305–311.
Boone JD, Dobbin ZC, Straughn JM Jr, Buchsbaum DJ (2015) Ovarian and cervical cancer patient derived xenografts: The past, present, and future. Gynecol Oncol 138 (2): 486–491.
Cairns RA, Hill RP (2004) A fluorescent orthotopic model of metastatic cervical carcinoma. Clin Exp Metastasis 21 (3): 275–281.
Chaudary N, Hedley DW, Hill RP (2011) Orthotopic xenograft model of cervical cancer for studying microenvironmental effects on metastasis formation and response to drug treatment. Curr Protoc Pharmacol Chapter 14 (Unit 14): 19.
Chaudary N, Jaluba K, Pintilie M, Hill RP (2015) Establishment of orthotopic primary cervix cancer xenografts. Methods Mol Biol 1249: 381–391.
Chaudary N, Pintilie M, Hedley D, Fyles AW, Milosevic M, Clarke B, Hill RP, Mackay H (2012b) Hedgehog pathway signaling in cervical carcinoma and outcome after chemoradiation. Cancer 118 (12): 3105–3115.
Chaudary N, Pintilie M, Hedley D, Fyles AW, Milosevic M, Clarke B, Hill RP, Mackay H (2014) Hedgehog signalling in primary cervix xenograft (OCICx) models: a potential new therapeutic target in combination with chemoradiotherapy. J Clin Oncol 32 (5s suppl): abstr 11120.
Chaudary N, Pintilie M, Schwock J, Dhani N, Clarke B, Milosevic M, Fyles A, Hill RP (2012) Characterization of the tumor-microenvironment in patient-derived cervix xenografts (OCICx). Cancers (Basel) 4 (3): 821–845.
Clarkson R, Lindsay PE, Ansell S, Wilson G, Jelveh S, Hill RP, Jaffray DA (2011) Characterization of image quality and image-guidance performance of a preclinical microirradiator. Med Phys 38 (2): 845–856.
Funke MGG, Silberstein PT (2015) Demographic and insurance-based disparities in diagnosis of stage IV cervical cancer: A population-based analysis using NCDB. J Clin Oncol 33 (suppl): e17589.
Gani C, Coackley C, Kumareswaran R, Schutze C, Krause M, Zafarana G, Bristow RG (2015) in vivo studies of the PARP inhibitor, AZD-2281, in combination with fractionated radiotherapy: An exploration of the therapeutic ratio. Radiother Oncol 116 (3): 486–494.
Greer BE, Koh WJ, Abu-Rustum NR, Apte SM, Campos SM, Chan J, Cho KR, Copeland L, Crispens MA, Dupont N, Eifel PJ, Gaffney DK, Huh WK, Kapp DS, Lurain JR 3rd, Martin L, Morgan MA, Morgan RJ Jr, Mutch D, Remmenga SW, Reynolds RK, Small W Jr, Teng N, Valea FA National Comprehensive Cancer N (2010) Cervical cancer. J Natl Compr Canc Netw 8 (12): 1388–1416.
Hanna A, Shevde LA (2016) Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer 15: 24.
Harrington KJ, Billingham LJ, Brunner TB, Burnet NG, Chan CS, Hoskin P, Mackay RI, Maughan TS, Macdougall J, McKenna WG, Nutting CM, Oliver A, Plummer R, Stratford IJ, Illidge T (2011) Guidelines for preclinical and early phase clinical assessment of novel radiosensitisers. Br J Cancer 105 (5): 628–639.
Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, Roman-Roman S, Seoane J, Trusolino L, Villanueva A (2014) Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 4 (9): 998–1013.
Hooper JE, Scott MP (2005) Communicating with hedgehogs. Nat Rev Mol Cell Biol 6 (4): 306–317.
Ingham PW, McMahon AP (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15 (23): 3059–3087.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61 (2): 69–90.
John T, Kohler D, Pintilie M, Yanagawa N, Pham NA, Li M, Panchal D, Hui F, Meng F, Shepherd FA, Tsao MS (2011) The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin Cancer Res 17 (1): 134–141.
Justilien V, Fields AP (2015) Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res 21 (3): 505–513.
Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, Chang WC, Kantoff E, Vannier MW, Catenacci DV, Venook AP, Kindler HL (2016) A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 45 (3): 370–375.
Ludwig JA, Weinstein JN (2005) Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer 5 (11): 845–856.
Ma CM, Coffey CW, DeWerd LA, Liu C, Nath R, Seltzer SM, Seuntjens JP American Association of Physicists in M (2001) AAPM protocol for 40–300 kV x-ray beam dosimetry in radiotherapy and radiobiology. Med Phys 28 (6): 868–893.
Meng E, Hanna A, Samant RS, Shevde LA (2015) The impact of hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers (Basel) 7 (3): 1333–1348.
Meerang M, Berard K, Felley-Bosco E, Lauk O, Vrugt B, Boss A, Kenkel D, Broggini-Tenzer A, Stahel RA, Arni S, Weder W, Opitz I (2016) Antagonizing the hedgehog pathway with vismodegib impairs malignant pleural mesothelioma growth in vivo by affecting stroma. Mol Cancer Ther 15 (5): 1095–1105.
O'Reilly KE, de Miera EV, Segura MF, Friedman E, Poliseno L, Han SW, Zhong J, Zavadil J, Pavlick A, Hernando E, Osman I (2013) Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel) 6 (11): 1429–1450.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324 (5933): 1457–1461.
Onishi H, Katano M (2014) Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol 20 (9): 2335–2342.
Pasca di Magliano M, Hebrok M (2003) Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 3 (12): 903–911.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Scales SJ, de Sauvage FJ (2009) Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci 30 (6): 303–312.
Scales SJ, de Sauvage FJ (2009) Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci 30 (6): 303–312.
Shevde LA, Samant RS (2014) Nonclassical hedgehog-GLI signaling and its clinical implications. Int J Cancer 135 (1): 1–6.
Statistics CC (2015) www.wcrf.org/int/cancer-facts-figures/data-specific-cancers/cervical-cancer-statistics. In World Cancer Research Fund International.
Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM, Alvarez RD, Landen CN (2012) Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol Cancer Ther 11 (7): 1587–1597.
Teichman J (2012) Hedgehog signalling and tumour-initiating cells as radioresistance factors in esophageal adenocarcinoma. Master of Science, Graduate Department of Medical Biophysics, University of Toronto, Available at https://tspace.library.utoronto.ca/bitstream/1807/33560/3/Teichman_Jennifer_R_201211_MSc_thesis.pdf.
Tucker SL, Withers HR, Mason KA, Thames HD Jr (1983) A dose-surviving fraction curve for mouse colonic mucosa. Eur J Cancer Clin Oncol 19 (3): 433–437.
van Hoof SJ, Granton PV, Verhaegen F (2013) Development and validation of a treatment planning system for small animal radiotherapy: SmART-Plan. Radiother Oncol 109 (3): 361–366.
Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H (2015) Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res 21 (15): 3561–3568.
Withers HR, Elkind MM (1970) Microcolony survival assay for cells of mouse intestinal mucosa exposed to radiation. Int J Radiat Biol Relat Stud Phys Chem Med 17 (3): 261–267.
Withers HR, Mason K, Reid BO, Dubravsky N, Barkley HT Jr, Brown BW, Smathers JB (1974) Response of mouse intestine to neutrons and gamma rays in relation to dose fractionation and division cycle. Cancer 34 (1): 39–47.
Withers HR, Mason KA (1974) The kinetics of recovery in irradiated colonic mucosa of the mouse. Cancer 34 (Suppl 3): S896–S903.
Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL, de Sauvage FJ (2008) A paracrine requirement for hedgehog signalling in cancer. Nature 455 (7211): 406–410.
Acknowledgements
Michael and Libby Goldgrub Foundation and the Canadian Association of Radiation Oncology RAZCER Research Award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Chaudary, N., Pintilie, M., Hedley, D. et al. Hedgehog inhibition enhances efficacy of radiation and cisplatin in orthotopic cervical cancer xenografts. Br J Cancer 116, 50–57 (2017). https://doi.org/10.1038/bjc.2016.383
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.383
Keywords
This article is cited by
-
Cancer-associated fibroblasts: a versatile mediator in tumor progression, metastasis, and targeted therapy
Cancer and Metastasis Reviews (2024)
-
The role of Hedgehog and Notch signaling pathway in cancer
Molecular Biomedicine (2022)
-
miR-636 inhibits EMT, cell proliferation and cell cycle of ovarian cancer by directly targeting transcription factor Gli2 involved in Hedgehog pathway
Cancer Cell International (2021)
-
Exosomes from cervical cancer cells facilitate pro-angiogenic endothelial reconditioning through transfer of Hedgehog–GLI signaling components
Cancer Cell International (2021)
-
Nuclear receptor corepressor (NCoR) is a positive prognosticator for cervical cancer
Archives of Gynecology and Obstetrics (2021)
