
Abstract
Background:
Clinical options for patients harbouring advanced/recurrent uterine serous carcinoma (USC), an aggressive variant of endometrial tumour, are very limited. Next-generation sequencing (NGS) data recently demonstrated that cyclin E1 (CCNE1) gene amplification and pik3ca driver mutations are common in USC and may therefore represent ideal therapeutic targets.
Methods:
Cyclin E1 expression was evaluated by immunohistochemistry (IHC) on 95 USCs. The efficacy of the cyclin-dependent kinase 2/9 inhibitor CYC065 was assessed on multiple primary USC cell lines with or without CCNE1 amplification. Cell-cycle analyses and knockdown experiments were performed to assess CYC065 targeting specificity. Finally, the in vitro and in vivo activity of CYC065, Taselisib (a PIK3CA inhibitor) and their combinations was tested on USC xenografts derived from CCNE1-amplified/pik3ca-mutated USCs.
Results:
We found that 89.5% of the USCs expressed CCNE1. CYC065 blocked cells in the G1 phase of the cell cycle and inhibited cell growth specifically in CCNE1-overexpressing USCs. Cyclin E1 knockdown conferred increased resistance to CYC065, whereas CYC065 treatment of xenografts derived from CCNE1-amplified USCs significantly reduced tumour growth. The combination of CYC065 and Taselisib demonstrated synergistic effect in vitro and was significantly more effective than single-agent treatment in decreasing tumour growth in xenografts of CCNE1-amplified/pik3ca-mutated USCs.
Conclusions:
Dual CCNE1/PIK3CA blockade may represent a novel therapeutic option for USC patients harbouring recurrent CCNE1-amplified/pi3kca-mutated tumours.
Similar content being viewed by others

Main
Uterine serous carcinoma (USC) constitutes only 10% of all endometrial cancers, but accounts for a disproportionate number of deaths among women affected with endometrial tumours (Siegel et al, 2015). Currently, the clinical management of advanced-stage USC includes cytoreductive surgery followed by systemic cytotoxic chemotherapy with or without localised radiation therapy (Hendrickson et al, 1982; Moore and Fader, 2011). Unfortunately, secondary to the intrinsic and/or rapid development of resistance to chemotherapy most advanced stage USC patients develop recurrent disease, which is fatal in the majority of the cases (Goff et al, 1994). Clinical options for the treatment of recurrent chemotherapy-resistant disease are very limited (Schwartz, 2006). Novel, more effective treatment modalities for the management of recurrent USC are desperately needed.
The genomic landscape of endometrial carcinoma has recently been evaluated by the Cancer Genome Atlas (TCGA) Research Network using multiple sequencing-based technologies (Cancer Genome Atlas Research Network et al, 2013). Data revealed that serous and serous-like endometrioid tumours have a unique genomic profile among endometrial cancers, characterised by frequent TP53 mutations and extensive copy number alterations (Cancer Genome Atlas Research Network et al, 2013). Importantly, the TCGA data also showed that ∼85% of these tumours exhibited a significant deregulation in the expression of cell-cycle-related genes (Cancer Genome Atlas Research Network et al, 2013). Consistent with these results, a comprehensive next-generation sequencing (NGS) evaluation of 57 USCs from our research group has recently shown that alterations in the cell-cycle pathway occurred in up to 88% of these tumours (Zhao et al, 2013). Remarkably, the amplification of the gene encoding for cyclin E1 (CCNE1) was reported in 48% of the samples analysed.
Cyclin E1 is a critical regulator of the G1–S phase transition in mammalian cells. Following binding to cyclin-dependent kinase 2 (CDK2), the CCNE1/CDK2 kinase complex is responsible for the regulation of several important S-phase processes including retinoblastoma (Rb) phosphorylation, histone biosynthesis and prereplication complex assembly (Reed, 1996; Ekholm and Reed, 2000; Hubalek et al, 2004). Upregulation of CCNE1 has been reported in many types of human tumours including high-grade serous ovarian and breast cancers and has been shown to correlate with worse prognosis (Sui et al, 2001; Rosen et al, 2006; Scaltriti et al, 2011; Lundgren et al, 2015). Although these evidences identify CCNE1 as an attractive therapeutic target in multiple tumour types, developing molecules to specifically target CCNE1 have so far been difficult because CCNE1 operates as a regulatory subunit of CDK2 rather than as a kinase or a receptor. The use of CDK2 inhibitors to indirectly interfere with CCNE1 function has been therefore proposed as an alternative approach in the past years (Asghar et al, 2015; Taylor-Harding et al, 2015; Yang et al, 2015). Accordingly, the antitumour activity of CYC065 (Cyclacel Ltd, Dundee, UK), a second-generation, orally available ATP-competitive inhibitor of CDK2/9 kinases has been previously evaluated on CCNE1-amplified breast cancer cells (Scaltriti et al, 2011). Results showed that CYC065 is able to significantly inhibit tumour growth in xenografts of CCNE1-amplified breast cancer. Importantly, this study identified CCNE1 amplification as one of the acquired mechanism of resistance to Trastuzumab, a monoclonal antibody targeting Her2/neu, and demonstrated that the treatment of Trastuzumab-resistant/CCNE1-amplified tumour cells with CYC065 was able to overcome such resistance, suggesting a molecular connection between CCNE1 and the Her2/PI3K/AKT/mTOR signalling pathways. Of interest, USC is frequently characterised by the hyperactivation of the Her2/PI3K/AKT/mTOR pathway (Cancer Genome Atlas Network, 2012, 2013; Zhao et al, 2013) and recent studies by our group showed that the presence of oncogenic mutations in the Pik3ca gene may not only confer resistance to Trastuzumab but also sensitise USC primary cell lines to the PIK3CA inhibitors such as Taselisib (English et al, 2013; Black et al, 2015; Lopez et al, 2015).
In this study, we first evaluated CCNE1 expression in USC by IHC using a tissue microarray (TMA) including 95 samples and then tested the in vitro and in vivo effectiveness of the CDK2/9 inhibitor CYC065 against multiple primary USC cell lines in vitro. Next, we evaluated the in vitro and in vivo activity of CYC065 and Taselisib as single agents or in combination against USCs harbouring amplification of CCNE1 and activating mutations in the Her2/PI3K/AKT/mTOR pathway. We demonstrate for the first time that the dual targeting of CCNE1 and PIK3CA with CYC065 and Taselisib is synergistic against CCNE1-amplified/pik3ca-mutated USC primary cell lines in vitro as well as in vivo. These results may have important implications for USC patients harbouring chemotherapy-resistant/recurrent disease.
Materials and Methods
Tissue microarray cyclin E immunohistochemistry
Study approval was obtained from the Ethical Institutional Review Board at Yale University according to the institutional guidelines. Tissue microarray was constructed using archived formalin-fixed, paraffin-embedded surgical pathology blocks from 95 previously diagnosed USC cases at the Department of Pathology, Yale University. Each tumour was represented in duplicates of 0.6 mm cores on the TMA. Cyclin E1 immunohistochemistry was performed on 4 μm TMA sections. Following deparaffinisation and rehydration, endogenous peroxidase was blocked in 3% H2O2. Steam and high pH (pH 9) were used for antigen retrieval. The slides were then incubated overnight at 4 °C with cyclin E antibody (clone HE12, 1 : 400 dilution; Millipore, Billerica, MA, USA). EnVision system (Dako, Carpinteria, CA, USA) was used for secondary detection and the reactions were visualised with diaminobenzidine. Appropriate positive and negative controls were used. Only nuclear immunoreactivity was considered positive. Immunostaining was assessed using a semiquantitative scoring system, as follows: 0, negative (<5% of tumour cell nuclei staining); 1, weakly/focally (5–50% of tumour nuclei) positive; 2, strongly/diffusely (>50% of tumour cell nuclei) positive.
Inhibitors
Taselisib was purchased from Medchemexpress (Monmouth Junction, NJ, USA), whereas CYC065 was provided by Cyclacel. Both compounds were dissolved in DMSO as a 10 mM stock solution and diluted in culture medium immediately before use.
Cell viability assay and synergism
The effect of CYC065 on the viability and IC50 of USC-ARK-1, USC-ARK-2, USC-ARK-7, USC-ARK-4 and USC-ARK-6 USC primary cell lines was determined in flow-cytometry assays as described previously (Lopez et al, 2015). Briefly, tumour cells were plated in six-well plates and treated with a titration of CYC065 concentrations (i.e., ranging from 100 to 500 nM). After 72 h, cells were harvested, washed and stained with propidium iodide (PI; 5 μg ml−1) for flow cytometric counts. The percentage of viable cells was then normalised considering the vehicle-treated cells as 100% viable. Half-maximal inhibitory concentration values were determined using GraphPad Prism5 version 6 (GraphPad Prism software, San Diego, CA, USA). For drug combination studies, USC-ARK-1 and USC-ARK-2 cell lines were incubated with the combination of Taselisib and CYC065 at multiple paired concentrations including the IC50, the IC50/2 and the IC50*2 of each cell line to the corresponding drug (i.e., 10 nM of Taselisib and 198 nM of CYC065 for USC-ARK-1 and 50 nM of Taselisib and 62.5 nM of CYC065 for USC-ARK-2). Synergism was assessed by the combination index (CI), according to the mathematical method described by Chou and Talalay. CI values <1 define a synergistic activity of the combination treatment (Chou, 2006). The CI values were calculated using the CompuSyn software (ComboSyn, Paramus, NJ, USA).
Cell-cycle analysis
Uterine serous carcinoma cells were seeded in six-well tissue culture plates and 24 h later were treated with 100 nM of CYC065. After 48 h, treated and control cells were permeabilised with ice-cold 70% ethanol and fixed for 30 min at 4 °C. After spinning at 2000 r.p.m. for 5 min and discarding the supernatant, cells were resuspended in PBS1*. After additional spinning at 2000 r.p.m. for 5 min, 100 μl ribonuclease (100 μg ml−1, DNase free; Sigma, St Louis, MO, USA) was added for 5 min incubation at room temperature, before exposure to PI (50 μg ml−1). CYC065-treated and -untreated control cells were acquired with FACSCalibur, using Cell Quest software (BD Biosciences, San Jose, CA, USA) and were analyzed using the FlowJo software (Ashland, OR, USA).
Knockdown of CCNE1
USC-ARK-2 cell line was chosen as a representative CCNE1-amplified cell line to be tested in knockdown experiments. Cyclin-E1-specific siRNA oligonucleotides (i.e., CCNE1: 5′-GGAUGUUGACUGCCUUGAATT-3′ (sense) and 5′-UUCAAGGCAGUCAACAUCCAG-3′ (antisense)) and nonspecific MOCK siRNA duplexes were used as negative controls (purchased from Ambion, Waltham, MA, USA). Briefly, USC-ARK-2 cells were cultured in six-well plates and transfected with anti-CCNE1 siRNA duplexes at 10 nM in conjunction with 5 μl Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Mock transfections were used as negative controls. Uterine serous carcinoma cells were treated 24 h later with scalar doses of CYC065. After 72 h, cells were harvested and used either for RNA extraction or for cell viability assays.
Immunoblotting
Cells were seeded in Petri dishes and left to adhere overnight. The following day, cells were incubated with Taselisib (10 nM), CYC065 (200 nM) or the combination for 6 h. Cells were then scraped on ice and lysed with RIPA buffer supplemented with protease inhibitor cocktail as described previously (Lopez et al, 2015). Protein concentration was quantified with a BCA Protein Assay Kit (no. 23225; Thermo Scientific, Waltham, MA, USA), and 40 μg of protein lysates were loaded for SDS–PAGE using precast gradient gels (no. 456-1094; Bio-Rad, Hercules, CA, USA). Antibodies used were HER2/neu (no. 06-562; Millipore, Billerica, MA, USA), pHER2/neu (#2247S; Cell Signalling, Danvers, MA, USA), Rb (#554136; BD Pharmingen, San Jose, CA, USA), pRb (no. 9307S; Cell Signalling), CCNE1 (no. 05-363; Millipore), S6 (no. 2217S; Cell Signalling), pS6 (no. 4856; Cell Signalling) and GAPDH (no. 2118; Cell Signalling). Incubation with primary antibodies was carried overnight in 5% BSA or milk in TBS-Tween at 4 °C. After incubation, membranes were washed in TBS-Tween three times before being incubated for 1 h at room temperature with the anti-mouse or anti-rabbit HRP-linked secondary antibodies (anti-rabbit no. 7074S (Cell Signalling) and anti-mouse no. sc-2005 (Santa Cruz, Dallas, TX, USA)). After three additional washes, blots were developed using an enhanced chemiluminescent system (GEL Logic 1500; Carestream Health, Rochester, NY, USA).
In vivo therapy
The in vivo efficacy of CYC065 used as a single agent was evaluated on xenograft mouse models derived from the CCNE1-amplified USC-ARK-2 USC cell line after study protocol approval by the Institutional Animal Care and Use Committees (IACUC). Xenografts derived from the CCNE1-amplified, PIK3CA-mutated USC-ARK-1 cell line were used for evaluating the in vivo combination of CYC065 and Taselisib. Briefly, 5–7-week-old SCID mice (Harlan Laboratories, Indianapolis, IN, USA) were injected into the subcutaneous region with USC cells. A minimum of five animals per group were used. Treatments were administrated by oral gavage starting 1 week after tumour implantation when the size of the tumour was 0.125–0.150 cm3. Uterine serous carcinoma-ARK-2-derived xenografts were divided into two groups: one group of animal received the vehicle, whereas the experimental group received CYC065 (22.5 mg kg−1 daily for 3 weeks). Uterine serous carcinoma-ARK-1-derived xenografts were instead divided into four groups: one group received the vehicle (0.5% methylcellulose–0.2% Tween-80), one group received CYC065 (22.5 mg kg−1 daily for 3 weeks), one group received Taselisib (10mg kg−1 daily, 5 days per week per 3 weeks) and the last group received the combination of CYC065 and Taselisib. The size of the tumour at the initiation of treatment was 0.125–0.150 cm3. Mouse weight and tumour size was recorded two times a week for the entire experimental period. Tumour volume was calculated by the formula: V=length × (width)2/2 and was plotted as mean±s.e.m. Mice were killed according to the rules and regulations set forth by the IACUC at Yale. In vivo experiments were repeated three times.
Statistical analysis
Statistical analysis was performed using GraphPad Prism5 version 6 (GraphPad Software). In the viability assay experiments, the number of viable cells after treatment with CYC065 or Taselisib was normalised to the vehicle-treated control considered as 100% viable. Data were then fit via nonlinear regression to a normalised logistic curve against the base-10 logarithms of dose in molarity (M). The resulting parameters were used for the calculation of the IC50s. One-way ANOVA was used to determine the statistical significance of the effect of the combination of CYC065 and Taselisib in comparison with each of the single agents. Unpaired t-test was also used to evaluate significant differences in the tumour volumes at specific time points in the in vivo experiments. Error bars represent s.e.m. P<0.05 was considered statistically significant.
Results
Cyclin-E1 transcript levels correlate with CNV analysis
A recent NGS study by our group evaluated copy number variations (CNV) on 25 matched (tumour and normal tissue from the same patient) USC samples (Zhao et al, 2013). In agreement with the TCGA data in serous endometrial cancer, we found focal amplification of the CCNE1 gene on chromosome 19 in 48% of the USC cases (Supplementary Figure 1A). To assess whether the amplification of CCNE1 correlated with a higher expression of the CCNE1 transcript, the same whole-exome-sequenced USC samples described above were tested by real-time PCR. We found that USC samples with CCNE1 gain of function by CNV expressed significantly higher levels of CCNE1 transcript compared with USC samples found with copy neutral or loss of function of the gene (CCNE1-wt/loss USC; Supplementary Figure 1B; P=0.0013).
Tissue microarray immunohistochemistry and FISH results
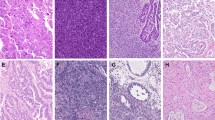
Next, we used IHC to evaluate the expression of CCNE1 at the protein level in 95 primary USC samples using a TMA. Cyclin E1 expression levels were scored from 0 to 2+, where 0 represented no staining, 1+ weak/focal staining and 2+ strong/diffuse staining. We found CCNE1 expression in 85 samples (89.5% of the total), with 33 (34.7%) of the USC demonstrating a score 1+ and 52 (54.8%) a score 2+. Of the CCNE1+ samples, 38 (44.7%) were from early-stage (stages I and II) patients, whereas 47 (55.3%) were from advanced-stage (stages III and IV) patients. None of the patients received neoadjuvant chemotherapy before tissue collection. Of the 10 samples in which CCNE1 staining was not detected (0 score) (10.5% of the total), 9 (90%) belonged to patients harbouring early-stage (stages I and II) disease. Representative IHC pictures of 0, a 1+ and a 2+ USC are depicted in Figure 1.

Cyclin E1 expression by IHC in USC. Representative immunohistochemical staining for CCNE1 on USC samples scoring 0 (A), 1+ (B) and 2+ (C). A representative negative control (i.e., TMA USC sample stained without the primary antibody, D) and a representative positive control (i.e., cell block from a cell line (ARK2) with known CCNE1 amplification by fluorescent in situ hybridisation (FISH), E) are also shown. (All images at × 200 original magnification.)
Cyclin E1 overexpressing primary USC cell lines respond to the CDK2 inhibitor CYC065 in vitro
To evaluate whether the overexpression of CCNE1 correlated with sensitivity to the CDK2/9 inhibitor CYC065, five USC primary cell lines were selected for in vitro proliferative and molecular assays based on their similar growth rate and differential expression of CCNE1 tested by real-time PCR, FISH and western blot (Table 1 and Supplementary Figures 2 and 3). We consistently found USC cell lines expressing high CCNE1 mRNA and protein levels to be significantly more sensitive to treatment with CYC065 in vitro when compared with low CCNE1-expressing cell lines (Figure 2A and B; IC50: mean±s.d.=124.1±57.8 nM in CCNE1-overexpressing USC cell lines vs 415±117.5 nM in CCNE1 low expressors, respectively; P=0.0003). Importantly, low concentrations of CYC065 (i.e., 100 nM) caused an arrest in the G1 phase of the cell cycle only in the CCNE1-overexpressing USC cell lines (i.e., USC-ARK-2, USC-ARK-7) (Figure 2C).

Sensitivity of selected USC primary cell lines to CYC065. (A) Representative dose–response curves of two CCNE1-amplified primary USC cell lines (upper panel) and two primary USC cell lines without CCNE1 amplification (lower panel) following treatment with scalar doses of CYC065. (B) Scatter plot showing half-maximal inhibitory concentration (IC50) for CCNE1-amplified USC cell lines compared with USC cell lines without CCNE1 amplification. Cyclin-E1-amplified USC cell lines were significantly more sensitive to CYC065 in vitro compared with those without CCNE1 amplification (IC50: mean±s.d.=124.1±57.8 nM in the CCNE1-overexpressing USC cell lines and 415±117.5 nM in CCNE1 low expressors, respectively; P=0.0003). (C) Cell-cycle analysis following treatment of USC cell lines with 100 nM of CYC065. Incubation of cells with CYC065 caused an arrest in the G1 phase of the cell-cycle specifically in CCNE1-amplified USC cell lines (ARK-2 and ARK-7). (D) Cyclin-E1 knockdown resulted in a 9.29-fold increase in the IC50 compared with the control (Ctrl; cells transfected with MOCK) after treatment with scalar doses of CYC065 (P=0.021). (E) Daily treatment of USC-ARK-2-derived xenografts with 22.5 mg kg−1 of CYC065 significantly inhibited tumour growth compared with vehicle-treated mice (P=0.012 starting at day 9 of the treatment). No weight loss was reported during the treatment period (F).
In recent studies, the 6–8 h pulse treatment with CYC065 at 500–1000 nM has been reported to rapidly induce cell death via CDK9 inhibition in acute myeloid leukaemia (AML) and breast cancer cell lines (MacKay et al, 2015; Saladino et al, 2015). Consistently, we observed about two-fold increase in the percentage of cells expressing annexin V/PI (markers of early and late apoptosis, respectively) following 6 h treatment of the USC-ARK-7 primary cancer cell line with the same concentrations of CYC065 in vitro (fold increase annexin V/PI-positive cells=1.78±0.3 and 2.25±0.7 after treatment of cells with 500 and 1000 nM of CYC065, respectively; Supplementary Figure 4).
Knockdown of CCNE1 confers resistance to CYC065 in vitro
Next, to determine the specificity of CYC065 in targeting CCNE1 in the overexpressing USC cell lines, the expression of CCNE1 in USC-ARK-2 was knocked down using siRNA technology. Cyclin E1 knockdown resulted in a 9.29-fold increase in CYC065 IC50 compared with the control (cells transfected with MOCK) following treatment with scalar concentrations of the drug (Figure 2D, P=0.021). Interestingly, we found that CCNE1 knockdown caused a significant inhibition of tumour cells’ growth compared with MOCK-transfected cells even without treatment (Supplementary Figure 5; % of growth inhibition: mean±s.d.=49.5±14.4, P=0.027). These data suggest a high dependency of USC to CCNE1 for cell proliferation.
CYC065 inhibits tumour growth in xenografts established from CCNE1-amplified USC
To evaluate the therapeutic potential of CYC065 as a single agent, USC-ARK-2-derived xenografts were treated daily with CYC065 (22.5 mg kg−1) for a 3-week period. Tumour size and mouse weight were recorded two times a week. The daily administration of CYC065 resulted in a significant reduction of tumour growth compared with the vehicle-treated mice (Figure 2E, P=0.012 starting at day 9 of the treatment). No significant weight loss was reported during the entire treatment period (Figure 2F).
Synergistic effect of the combination of CYC065 and Taselisib in vitro
Because a recent NGS analysis from our group (Zhao et al, 2013) demonstrated that up to 91.7% of CCNE1-amplified USC may also carry mutations in genes involved in the Her2/PI3K/AKT/mTOR pathway (Supplementary Table 1), we next tested the activity of CYC065 and Taselisib alone or in combination using two USC primary tumour models (i.e., USC-ARK-1, a CCNE1/c-erbB2/pik3ca-amplified and pik3ca-mutated cell line and USC-ARK-2, a CCNE1/c-erbB2/pik3ca-amplified cell line harbouring wild-type PIK3CA gene; Supplementary Table 1). As shown in Figure 3A, the incubation of USC-ARK-1 and USC-ARK-2 cells with the combination of CYC065 and Taselisib was significantly more effective in inhibiting tumour growth than each of the single agents in both cell lines (Figure 3A, % viable cells: mean±s.d.=65.8±7.9, 75.4±11.3 and 48±7.6 for the CYC065, Taselisib and the combination, respectively, for USC-ARK-1; P<0.05), and 48.9±4.6, 36.7±5.5 and 15.9±4.05 for CYC065, Taselisib and the combination, respectively, for USC-ARK-2; P<0.05). Importantly, when we evaluated the effect of the combination of CYC065 and Taselisib at multiple paired concentrations using the CompuSyn software, we found a synergistic activity of the combination of CYC065 and Taselisib in both USC models (CI values: 0.93, 0.81, 0.71, 0.65 for Fa=0.5, 0.75, 0.9 and 0.95, respectively, in USC-ARK-1 and 0.45, 0.46, 0.54 and 0.64 for Fa=0.5, 0.75, 0.9 and 0.95, respectively, in USC-ARK-2).

Effect of the combination of CYC065 and Taselisib in vitro. (A) Uterine serous carcinoma-ARK-1 and USC-ARK-2 were treated with the half-maximal inhibitory concentration (IC50) of CYC065, the IC50 of Taselisib (GDC0032) or the combination of the IC50s of the two compounds for 72 h. Cell counting was then carried as described in Materials and Methods. The incubation of USC-ARK-1 and USC-ARK-2 cells with the combination of CYC065 and Taselisib was significantly more effective in inhibiting cells’ growth than each of the single agents (P<0.05). (B) Representative western blot analysis depicting changes in the expression levels of proteins involved in the Her2/PI3K/AKT/mTOR pathway (i.e., Her2/neu, pHer2/neu, S6 and pS6) and in the CCNE1 pathway (Rb, pRb and CCNE1) following the treatment of USC-ARK-1 and USC-ARK-2 with CYC065 (CYC), Taselisib (GDC) or the combination (combo) for 6 h. The combination of CYC065 and Taselisib was highly effective in inhibiting pRb and pS6 in both cell lines. CTRL, control.
Immunoblot analysis after combination treatment with CYC065 and Taselisib
Western blot analysis was carried out to investigate the molecular basis of the drug synergism after treatment of USC-ARK-1 and USC-ARK-2 with CYC065 and Taselisib. Changes in the total Rb, HER2/neu and S6 and in their phosphorylation status were evaluated after 6 h of treatment with 200 nM of CYC065, 10 nM of Taselisib or the combination. Changes in the expression of CCNE1 were also examined. As shown in Figure 3B, incubation with Taselisib was able to induce a decrease in the phosphorylation of S6 in both cell lines, whereas CYC065 was effective in inhibiting Rb phosphorylation in USC-ARK-1 cell line but not in USC-ARK-2. Importantly, the combination of CYC065 and Taselisib was highly effective in inhibiting pRb and pS6 in both cell lines (Figure 3B). No changes in total HER2/neu, pHER2/neu and total S6 were observed. Conversely, decreased total Rb was reported in USC-ARK-1 but not in USC-ARK-2 following treatment with the combination of CYC065 and Taselisib. An increased expression of CCNE1 was also observed after incubation with CYC065 and with the combination of CYC065 and Taselisib in both USC cell lines.
In vivo activity of CYC065 and Taselisib on USC xenografts
To assess whether the synergism observed in our in vitro assays also resulted in an improved therapeutic effect in vivo, Scid mice bearing USC-ARK-1-derived xenografts were treated with CYC065 (q.d. for 3 weeks), Taselisib (5 days a week for 3 weeks) or with the combination of the two drugs. As shown in Figure 4A, the combination of CYC065 and Taselisib was significantly more effective than each of the single agents in inhibiting tumour growth (mean±s.e.m. in tumour size at day 21 of treatment=1.04±0.5, 0.72±0.29 and 0.37±0.11 cm3 in the CYC065, in the Taselisib and in the combination groups, respectively; P<0.03). CYC065 used as single agent significantly inhibited tumour growth of USC-ARK-1-derived xenografts compared with vehicle-treated mice (mean±s.e.m. in tumour size at day 17 of treatment: 1.29±0.12 and 0.80±0.34 cm3 in the control and in the CYC065 group, respectively; P=0.028). The weight of the animals in the different groups was monitored two times a week during the course of the experiment. No significant differences were reported between the different groups, although a slight decrease in mice weight was observed in the combination group starting from day 19 of the treatment (Figure 4B; P>0.05).

Effect of the combination of CYC065 and Taselisib on tumour growth in vivo. Scid mice bearing USC-ARK-1-derived xenografts were treated with CYC065 (q.d. (one a day) for 3 weeks), Taselisib (5 days a week for 3 weeks) or with the combination (combo) of the two drugs as described in Materials and Methods. The combination of CYC065 and Taselisib was more effective than each of the single agents in inhibiting tumour growth. (A) Mean±s.d. in tumour size at day 21 of treatment=1.04±0.5, 0.72±0.29 and 0.37±0.11 cm3 in CYC065, Taselisib and in the combination groups, respectively (P<0.03). No significant weight loss was reported in any of the experimental groups, although a slight decrease in mice weight was observed in the combination group starting from day 19 of the treatment (B: P>0.05).
Discussion
Uterine serous carcinoma is the most aggressive variant of endometrial cancer (Siegel et al, 2015). Owing to the intrinsic resistance of this tumour to chemotherapy, patients diagnosed with advanced-stage USC have an extremely poor prognosis (Schwab and Santin, 2015). The development of novel and more effective therapeutic strategies to target chemotherapy resistant/recurrent USC therefore remains of paramount importance.
Multiple comprehensive NGS studies performed by us as well as other groups have recently analysed the genetic landscape of USC (Kuhn et al, 2012, 2014; Cancer Genome Atlas Research Network et al, 2013; Zhao et al, 2013). The results of these studies consistently indicated that the amplification of the CCNE1 gene on chromosome 19 and mutations in the pik3ca gene on chromosome 3 represent two of the most frequent genetic alterations identified in USC. However, while CCNE1 may represent an attractive therapeutic target, to our knowledge, no studies have yet evaluated CCNE1 expression in a large cohort of USC specimens and, more importantly, validated CCNE1 as a novel biomarker in USC cell lines in in vitro and in vivo experiments.
Cyclin E1, an activator of CDK2, is known to have a critical role in the regulation of the G1–S phase transition in mammalian cells. Once formed, the CCNE1/CDK2 complex promotes the phosphorylation of several proteins involved in cell cycle progression (i.e., p27 and Rb), DNA replication (i.e., replication factors A and C) and histone biosynthesis (i.e., NPAT) (Reed, 1996; Ekholm and Reed, 2000; Ma et al, 2000; Okuda et al, 2000; Sever-Chroneos et al, 2001; Hubalek et al, 2004; Whittaker et al, 2004; Shapiro, 2006; Bose et al, 2013). Overexpression of CCNE1 has been associated with a broad spectrum of human malignancies and found to correlate with worse prognosis (Sui et al, 2001; Rosen et al, 2006; Scaltriti et al, 2011; Lundgren et al, 2015). On the basis of these data, the use of CDK inhibitors (CDKi) to block the activity of the CCNE1/CDK2 complex as a therapeutic strategy for the treatment of multiple cancers has been extensively investigated over the past two decades (Asghar et al, 2015). First-generation CDKi (including alvocidib and seliciclib) tested in phase I and II clinical trials have shown limited potency or selectivity for CDK2 (Heath et al, 2008; Le Tourneau et al, 2010; Nemunaitis et al, 2013). Second-generation CDKi with varying CDKi profiles including increased potency against CDK2 are currently undergoing preclinical and/or clinical evaluation (Senderowicz et al, 1998; Taylor-Harding et al, 2015; Yang et al, 2015).
CYC065 is a recently developed second-generation CDK2/9 inhibitor with reported activity against AML and CCNE1-amplified breast cancer (Scaltriti et al, 2011; MacKay et al, 2015; Saladino et al, 2015). Accordingly, in the current study, we initially evaluated the expression of CCNE1 in USC by IHC in a large number of USC using a TMA. We found the majority of USC (89.5%) to express CCNE1 at moderate or high levels, strongly supporting a correlation between gene amplification, mRNA and protein expression results, and, importantly, suggesting that a large number of USC may be addicted to overactivation of the CCNE1 pathway. Interestingly, CCNE1 expression was found to be more prevalent in late stages (stages III and IV) USC, suggesting a preferential expression of this protein during the progression of the disease. On the basis of these results, we initially investigated the therapeutic potential of CYC065 against CCNE1-amplified USC cell lines as single agent. We found elevated CCNE1 expression to significantly correlate with sensitivity to CYC065 in vitro and that the treatment of USC cell lines with CYC065 caused accumulation of cells in the G1 phase of the cell cycle specifically in CCNE1-amplified USC cell lines. Knockdown of CCNE1 resulted in strong cell growth inhibition and in resistance to CYC065 supporting a relationship between high CCNE1 expression in USC and sensitivity to CYC065. These data are therefore in agreement with the results of other groups demonstrating an ‘oncogene addition’ of multiple human tumours including ovarian cancer (i.e., a tumour histologically similar to USC) to CCNE1 expression (Etemadmoghadam et al, 2010, 2013). Treatment of USC cell lines with higher concentrations of CYC065 also induced apoptosis as has been reported for CYC065 in other cancer cell lines (MacKay et al, 2015; Saladino et al, 2015). Importantly, CYC065 was effective in vivo in decreasing tumour growth in xenografts derived from CCNE1-amplified USC. Taken together, these data suggest a high dependency of USC cell proliferation on the CCNE1 pathway and promising activity of CYC065 in targeting the CCNE1-amplified USC cell lines both in vitro as well as in vivo.
Studies by our research group have recently shown that USC may be extremely sensitive in vitro to compounds that selectively target the Her2/PI3K/Akt/mTor pathway (English et al, 2013; Lopez et al, 2014, 2015; Schwab et al, 2014). In these studies, however, treatment with highly targeted PIK3CA agents was found to be only transiently effective in vivo in controlling the growth of USC xenograft models harbouring HER2/neu gene amplification or oncogenic pik3ca mutations. These in vivo data in animals therefore mirror long-term data in human patients where, after initial responses, progression is generally detected few months after starting targeted treatment in a large number of cancer patients (Engelman and Janne, 2008). Importantly, recent preclinical data strongly suggest that dual-targeting of HER2/PIK3CA with neratinib (a pan-c-erb inhibitor) and Taselisib may be highly synergistic and able to achieve durable regression of established USC xenografts in vivo (Lopez et al, 2015).
On the basis of these data and the recent observation that over 90% of CCNE1-amplified USC may also harbour oncogenic alterations in the genes encoding for proteins of the Her2/PI3K/AKT/mTOR pathway (i.e., pik3ca, c-erbB2, akt1), we next evaluated the activity of the combination of CYC065 and Taselisib against primary USC cell lines and tumour xenografts. We found the combined treatment to have a synergistic effect in inhibiting the growth of tumour cells in vitro in primary USC cell lines. We believe the synergism found to be at least in part the result of the simultaneous block of the CCNE1 and the PI3K pathways, two of the most frequently deregulated molecular pathways in USC (Kuhn et al, 2012; Cancer Genome Atlas Research Network et al, 2013; Zhao et al, 2013; Schwab and Santin, 2015). In support of this hypothesis, downstream PI3K and CCNE1 signalling assessed by immunoblotting experiments showed that the combination of Taselisib and CYC065 was highly effective in inhibiting the phosphorylation of S6 and Rb, the downstream effectors of PI3K and CCNE1/CDK2, respectively, and, importantly, significantly more efficacious than each of the single agents when used alone in inhibiting tumour growth in xenograft models derived from a CCNE1-amplified, pik3ca-mutated USC cell line.
As CDKi target proteins that are essential for the proliferation and the survival of normal as well as tumour cells, their use at high dose as single agents may be associated with significant toxicity (Senderowicz et al, 1998; Shapiro, 2006; Heath et al, 2008). A more rationale use of CDKi in USC patients might therefore be at a lower dose in combination with other molecularly targeted agents acting in synergy. Consistent with this view, Palbociclib, an inhibitor of CDK4 and 6, has recently been shown to be clinically effective (and for this reason approved by the FDA) when used in combination with letrozole for the treatment of postmenopausal women with oestrogen receptor-positive, Her2-negative advanced breast cancer (Beaver et al, 2015). Taken together, these data combined with our results strongly support the view that CYC065 may represent a promising novel compound to be considered either alone or in combination with selective Her2/PI3K/Akt/mTor inhibitors for the treatment of USC patients harbouring chemotherapy resistant/recurrent CCNE1-amplified endometrial cancer.
Change history
26 July 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 14 (2): 130–146.
Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, Zirkelbach JF, Yu J, Liu Q, Zhao L, Crich J, Chen XH, Hughes M, Bloomquist E, Tang S, Sridhara R, Kluetz PG, Kim G, Ibrahim A, Pazdur R, Cortazar P (2015) FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res 21 (21): 4760–4766.
Black JD, Lopez S, Cocco E, Bellone S, Altwerger G, Schwab CL, English DP, Bonazzoli E, Predolini F, Ferrari F, Ratner E, Silasi DA, Azodi M, Schwartz PE, Santin AD (2015) PIK3CA oncogenic mutations represent a major mechanism of resistance to trastuzumab in HER2/neu overexpressing uterine serous carcinomas. Br J Cancer 113 (11): 1641.
Bose P, Simmons GL, Grant S (2013) Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Invest Drugs 22 (6): 723–738.
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490 (7418): 61–70.
Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497 (7447): 67–73.
Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58 (3): 621–681.
Ekholm SV, Reed SI (2000) Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 12 (6): 676–684.
Engelman JA, Janne PA (2008) Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 14 (10): 2895–2899.
English DP, Bellone S, Cocco E, Bortolomai I, Pecorelli S, Lopez S, Silasi DA, Schwartz PE, Rutherford T, Santin AD (2013) Oncogenic PIK3CA gene mutations and HER2/neu gene amplifications determine the sensitivity of uterine serous carcinoma cell lines to GDC-0980, a selective inhibitor of Class I PI3 kinase and mTOR kinase (TORC1/2). Am J Obstet Gynecol 209 (5): 465 e1–465 e9.
Etemadmoghadam D, Au-Yeung G, Wall M, Mitchell C, Kansara M, Loehrer E, Batzios C, George J, Ftouni S, Weir BA, Carter S, Gresshoff I, Mileshkin L, Rischin D, Hahn WC, Waring PM, Getz G, Cullinane C, Campbell LJ, Bowtell DD (2013) Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin Cancer Res 19 (21): 5960–5971.
Etemadmoghadam D, George J, Cowin PA, Cullinane C, Kansara M Australian Ovarian Cancer Study G Gorringe KL, Smyth GK, Bowtell DD (2010) Amplicon-dependent CCNE1 expression is critical for clonogenic survival after cisplatin treatment and is correlated with 20q11 gain in ovarian cancer. PLoS One 5 (11): e15498.
Goff BA, Kato D, Schmidt RA, Ek M, Ferry JA, Muntz HG, Cain JM, Tamimi HK, Figge DC, Greer BE (1994) Uterine papillary serous carcinoma: patterns of metastatic spread. Gynecol Oncol 54 (3): 264–268.
Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM (2008) A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Invest New Drugs 26 (1): 59–65.
Hendrickson M, Ross J, Eifel P, Martinez A, Kempson R (1982) Uterine papillary serous carcinoma: a highly malignant form of endometrial adenocarcinoma. Am J Surg Pathol 6 (2): 93–108.
Hubalek MM, Widschwendter A, Erdel M, Gschwendtner A, Fiegl HM, Muller HM, Goebel G, Mueller-Holzner E, Marth C, Spruck CH, Reed SI, Widschwendter M (2004) Cyclin E dysregulation and chromosomal instability in endometrial cancer. Oncogene 23 (23): 4187–4192.
Kuhn E, Bahadirli-Talbott A, Shih Ie M (2014) Frequent CCNE1 amplification in endometrial intraepithelial carcinoma and uterine serous carcinoma. Mod Pathol 27 (7): 1014–1019.
Kuhn E, Wu RC, Guan B, Wu G, Zhang J, Wang Y, Song L, Yuan X, Wei L, Roden RB, Kuo KT, Nakayama K, Clarke B, Shaw P, Olvera N, Kurman RJ, Levine DA, Wang TL, Shih Ie M (2012) Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst 104 (19): 1503–1513.
Le Tourneau C, Faivre S, Laurence V, Delbaldo C, Vera K, Girre V, Chiao J, Armour S, Frame S, Green SR, Gianella-Borradori A, Dieras V, Raymond E (2010) Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur J Cancer 46 (18): 3243–3250.
Lopez S, Cocco E, Black J, Bellone S, Bonazzoli E, Predolini F, Ferrari F, Schwab CL, English DP, Ratner E, Silasi DA, Azodi M, Schwartz PE, Terranova C, Angioli R, Santin AD (2015) Dual HER2/PIK3CA targeting overcomes single-agent acquired resistance in HER2-amplified uterine serous carcinoma cell lines in vitro and in vivo. Mol Cancer Ther 14 (11): 2519–2526.
Lopez S, Schwab CL, Cocco E, Bellone S, Bonazzoli E, English DP, Schwartz PE, Rutherford T, Angioli R, Santin AD (2014) Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol 135 (2): 312–317.
Lundgren C, Ahlin C, Holmberg L, Amini RM, Fjallskog ML, Blomqvist C (2015) Cyclin E1 is a strong prognostic marker for death from lymph node negative breast cancer. A population-based case–control study. Acta Oncol 54 (4): 538–544.
Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, Harper JW (2000) Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev 14 (18): 2298–2313.
MacKay C, Frame S, Saladino C, Pohler E, Zheleva D, Blake DG (2015) Abstract B182: Molecular basis for clinical development of the novel CDK2/9 inhibitor CYC065 in oncology. Mol Cancer Ther 14: B182.
Moore KN, Fader AN (2011) Uterine papillary serous carcinoma. Clin Obstet Gynecol 54 (2): 278–291.
Nemunaitis JJ, Small KA, Kirschmeier P, Zhang D, Zhu Y, Jou YM, Statkevich P, Yao SL, Bannerji R (2013) A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Transl Med 11: 259.
Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K (2000) Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103 (1): 127–140.
Reed SI (1996) Cyclin E: in mid-cycle. Biochim Biophys Acta 1287 (2–3): 151–153.
Rosen DG, Yang G, Deavers MT, Malpica A, Kavanagh JJ, Mills GB, Liu J (2006) Cyclin E expression is correlated with tumor progression and predicts a poor prognosis in patients with ovarian carcinoma. Cancer 106 (9): 1925–1932.
Saladino C, Frame S, Davis S, Blake D, Zheleva D (2015) Abstract 1650: CYC065, a novel CDK2/5/9 inhibitor: detailed mechanistic studies, determinants of sensitivity and synergistic combinations. Cancer Res 75: 1650–1650.
Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, Guzman M, Gili M, Rodriguez O, Rodriguez S, Perez J, Green SR, Mai S, Rosen N, Hudis C, Baselga J (2011) Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci USA 108 (9): 3761–3766.
Schwab CL, English DP, Roque DM, Bellone S, Lopez S, Cocco E, Nicoletti R, Rutherford TJ, Schwartz PE, Santin AD (2014) Neratinib shows efficacy in the treatment of HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol 135 (1): 142–148.
Schwab CL, Santin AD (2015) Targeted therapy in the treatment of uterine serous carcinoma. Pharmacogenomics 16 (2): 97–99.
Schwartz PE (2006) The management of serous papillary uterine cancer. Curr Opin Oncol 18 (5): 494–499.
Senderowicz AM, Headlee D, Stinson SF, Lush RM, Kalil N, Villalba L, Hill K, Steinberg SM, Figg WD, Tompkins A, Arbuck SG, Sausville EA (1998) Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol 16 (9): 2986–2999.
Sever-Chroneos Z, Angus SP, Fribourg AF, Wan H, Todorov I, Knudsen KE, Knudsen ES (2001) Retinoblastoma tumor suppressor protein signals through inhibition of cyclin-dependent kinase 2 activity to disrupt PCNA function in S phase. Mol Cell Biol 21 (12): 4032–4045.
Shapiro GI (2006) Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 24 (11): 1770–1783.
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65 (1): 5–29.
Sui L, Dong Y, Ohno M, Sugimoto K, Tai Y, Hando T, Tokuda M (2001) Implication of malignancy and prognosis of p27(kip1), Cyclin E, and Cdk2 expression in epithelial ovarian tumors. Gynecol Oncol 83 (1): 56–63.
Taylor-Harding B, Aspuria PJ, Agadjanian H, Cheon DJ, Mizuno T, Greenberg D, Allen JR, Spurka L, Funari V, Spiteri E, Wang Q, Orsulic S, Walsh C, Karlan BY, Wiedemeyer WR (2015) Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 6 (2): 696–714.
Whittaker SR, Walton MI, Garrett MD, Workman P (2004) The cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res 64 (1): 262–272.
Yang L, Fang D, Chen H, Lu Y, Dong Z, Ding HF, Jing Q, Su SB, Huang S (2015) Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 6 (25): 20801–20812.
Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, Bortolomai I, Buza N, Hui P, Abu-Khalaf M, Ravaggi A, Bignotti E, Bandiera E, Romani C, Todeschini P, Tassi R, Zanotti L, Carrara L, Pecorelli S, Silasi DA, Ratner E, Azodi M, Schwartz PE, Rutherford TJ, Stiegler AL, Mane S, Boggon TJ, Schlessinger J, Lifton RP, Santin AD (2013) Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci USA 110 (8): 2916–2921.
Acknowledgements
This work was supported, in part, by R01 CA154460-01 and U01 CA176067-01A1 grants from NIH, the Deborah Bunn Alley Foundation, the Tina Brozman Foundation, the Discovery to Cure Foundation and the Guido Berlucchi Foundation (to ADS). This investigation was also supported by NIH Research Grant CA-16359 from the NCI and by the Italian Ministry of Health Grant RF-2010-2313497 (to ADS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Cocco, E., Lopez, S., Black, J. et al. Dual CCNE1/PIK3CA targeting is synergistic in CCNE1-amplified/PIK3CA-mutated uterine serous carcinomas in vitro and in vivo. Br J Cancer 115, 303–311 (2016). https://doi.org/10.1038/bjc.2016.198
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.198
Keywords
This article is cited by
-
Integrative molecular and clinical profiling of acral melanoma links focal amplification of 22q11.21 to metastasis
Nature Communications (2022)
-
Cyclin-dependent kinase inhibitor fadraciclib (CYC065) depletes anti-apoptotic protein and synergizes with venetoclax in primary chronic lymphocytic leukemia cells
Leukemia (2022)
-
Invasive lobular carcinoma: an understudied emergent subtype of breast cancer
Breast Cancer Research and Treatment (2022)
-
Long noncoding RNA LINC01234 silencing exerts an anti-oncogenic effect in esophageal cancer cells through microRNA-193a-5p-mediated CCNE1 downregulation
Cellular Oncology (2020)
