
Abstract
Background:
Olaparib (AZD2281), a PARP-1/2 inhibitor, has been extensively investigated in clinical trials. However, limited clinical data are available about its long-term safety and anti-tumour activity.
Methods:
Patients had first participated in a phase I study of olaparib combined with carboplatin and/or paclitaxel. They continued with olaparib monotherapy in their best interest if they failed to tolerate the combination due to the treatment-related adverse events (TRAEs). Safety data were collected by physical examination and regular laboratory evaluations. Disease evaluations were performed by CT scan.
Results:
At data cutoff, 21 patients were included; 10 with breast, 9 with ovarian and 2 with fallopian tube cancer of whom 16 patients had a BRCA mutation (13 BRCA1; 3 BRCA2). TRAEs were mostly haematological and most prominent shortly after switching from combination to monotherapy, probably due to carry-over effects of chemotherapy. Over time, both severity and frequency of TRAEs decreased. Responses to olaparib were durable with a median treatment duration of 52 (range 7–183) weeks. In total, nine (43%) patients were still on study at data cutoff.
Conclusion:
Continued long-term daily olaparib was found to be safe and tolerable. Encouragingly, patients who showed a favourable response on earlier combination therapy maintained this response on olaparib monotherapy.
Similar content being viewed by others


Main
The enzyme poly (ADP-ribose) polymerase (PARP) is able to repair single-strand breaks (SSBs) in DNA. Accordingly, inhibiting PARP can cause SSBs to persist, leading to double-strand breaks (DSBs). The most error-free mechanism to repair these DSBs is via the homologous recombination (HR) pathway. Cells that are HR-deficient instead initiate alternative, more error-prone pathways to repair DSBs, such as non-homologous end joining or single-strand annealing. This can cause genomic instability, eventually leading to cell death (Ashworth, 2008; Sandhu et al, 2010; Dedes et al, 2011; Helleday, 2011; Yap et al, 2011). Thus, inhibiting PARP in HR-deficient tumour cells, such as those carrying BRCA mutations, is an effective treatment strategy, as evidenced in preclinical models (Bryant et al, 2005; Farmer et al, 2005). Olaparib (AZD2281; AstraZeneca, Macclesfield, UK) is one of the first known PARP inhibitors to exploit this so-called synthetic lethality, selectively inhibiting PARP-1 and PARP-2, both in vitro and in vivo (Menear et al, 2008). In a phase I study, the maximum tolerated dose (MTD) for monotherapy was 400 mg as capsules twice daily (BID). Main toxicities were mild and included nausea/vomiting, fatigue and a low incidence of myelosuppression. Olaparib also showed impressive anti-tumour activity, especially in BRCA1/2 mutation carriers, with 12 out of 19 (63%) patients with BRCA-mutated tumours deriving clinical benefit (Fong et al, 2009). As monotherapy, olaparib demonstrated anti-tumour activity in a phase II study in patients with high-grade serous ovarian cancer, with response rates being as high as 41% in BRCA1/2 mutation carriers (Gelmon et al, 2011). However, although a placebo-controlled phase II study in platinum-sensitive ovarian cancer patients conducted with olaparib as maintenance therapy after chemotherapy clearly showed an increase in progression-free survival (PFS) for olaparib (8.4 vs 4.8 months with placebo), the drug failed to induce prolonged overall survival and further development was put on hold (Bennet, 2012; Ledermann et al, 2012). A subsequent analysis showed an increase in PFS (11.2 vs 4.3 months) in BRCA mutation carriers compared with wild-type patients (Ledermann et al, 2013). Inhibition of a DNA-repair pathway could also sensitise tumour cells to the DNA-damaging effects of chemotherapies that cause SSBs. This hypothesis has been supported by preclinical models (Rottenberg et al, 2008) and has subsequently been tested in the clinic. In clinical trials where olaparib was combined with chemotherapy, it greatly enhanced chemotherapy-induced myelosuppression. Phase I studies that combined olaparib with dacarbazine, topotecan or cisplatin/gemcitabine all reported dose-limiting toxicities of thrombocytopenia, (febrile) neutropenia and anaemia (Khan et al, 2011; Rajan et al, 2012; Samol et al, 2012). Also, increased bone marrow toxicity were observed in clinical trials where olaparib was combined with carboplatin compared with carboplatin alone (Oza et al, 2015). This increase in myelosuppression could be attributed to an increase in the sensitivity of rapidly dividing bone marrow cells to the toxic effects of DNA-damaging therapies by olaparib (De Murcia et al, 1997). In a recent phase I study, in which olaparib was combined with carboplatin and paclitaxel, increased myelosuppression was also observed, requiring frequent dose modifications, including interruptions, delays and reductions. These toxicities and modifications appeared to be more frequent with increasing doses of olaparib (doses ranged from 50 to 400 mg BID; Van der Noll et al, 2013a, 2013b). However, patients who could not tolerate the combination but showed clinical benefit were given the option to continue olaparib as monotherapy after six cycles. In this subpopulation, safety and anti-tumour activity data of long-term administration of olaparib were prospectively collected. As PARP has different roles in the body (e.g., it is also implicated in inflammation, diabetes and ischaemia), it could be anticipated that inhibiting PARP over a long period of time might lead to previously unobserved adverse effects (Bürkle, 2001; Krishnakumar and Kraus, 2010; Sodhi et al, 2010). Although many clinical trials have been undertaken with olaparib, no study has previously described its long-term effects. In the current paper, we present data on long-term safety and anti-tumour activity of olaparib monotherapy after combination treatment with chemotherapy in patients with advanced breast, ovarian or fallopian tube cancer.
Materials and Methods
Patient selection and eligibility
This study was performed at The Netherlands Cancer Institute. In a previous phase I study, patients had received olaparib as capsules or tablets, continuously or discontinuously and in doses ranging from 50 to 200 mg BID in combination with carboplatin and/or paclitaxel. Patients treated in this phase I study were offered to continue with olaparib monotherapy in their best interest in case they had completed at least 6 cycles of combination treatment and experienced substantial toxicity without signs of disease progression. The patients included in this monotherapy trial came from both the dose escalation as the dose expansion parts. Eligibility criteria included the following: inability to continue olaparib in combination with carboplatin and/or paclitaxel due to persistent myelosuppression (neutro- and or thrombocytopenia CTC grade ⩾2), development of specific chemotherapy-related adverse events, such as sensory neurotoxicity or allergic reactions or patient request to stop combination treatment; age ⩾18 years; ECOG performance status ⩽2 and adequate bone marrow, hepatic and renal function (as evidenced by thrombocytes ⩾100 × 109 l−1; absolute neutrophil count ⩾1.5 × 109 l−1; haemoglobin ⩾6.2 mmol l−1; total bilirubin ⩽1.25 × upper normal limit (ULN); serum aspartate aminotransferase and alanine aminotransferase ⩽2.5 × ULN and serum creatinine ⩽1.5 × ULN).
Drug administration
Olaparib was administered in a capsule formulation to patients at the optimal recommended monotherapy dose of 400 mg BID – previously determined in the phase I monotherapy study – regardless of the dose level in which they had been included in the combination phase I trial. One treatment cycle lasted 28 days. If a patient experienced toxicity, olaparib administration could be interrupted according to the investigator’s discretion for up to a maximum of 28 days, until the toxicity reverted to ⩽grade 1. If toxicity did not resolve within 28 days, or recurred after re-challenge and/or multiple dose interruptions, a dose reduction was to be considered (first to 200 mg BID; second to 100 mg BID). If the toxicity persisted, treatment with olaparib was to be discontinued.
Safety evaluations
Patients had to undergo laboratory evaluations for haematology and chemistry at the start of a new cycle. The majority of patients underwent more frequent laboratory assessments during the first two cycles due to the persisting haematological toxicities after the combination treatment. Other safety evaluations – including physical examinations, measuring of vital signs and recording of adverse events and concomitant medications – were done at the start of every new cycle up to and including the sixth cycle. From then on these evaluations were done every other cycle up to cycle 12, and every three cycles thereafter. All adverse events were graded according to the Common Terminology Criteria for Adverse Events version 3.0 (Trotti et al, 2003). Grade 1 haematologic adverse events were not collected in this study, unless they led to clinically relevant interventions (such as blood transfusions for grade 1 anaemia).
Response assessments
To objectively assess the size of target lesions, CT or MRI scans were made every two cycles until cycle 12 and every three cycles thereafter, and evaluated according to Response Evaluation Criteria In Solid Tumours version 1.0 (Therasse et al 2000). Additional response evaluation was done by measuring relevant tumour markers, such as cancer antigen 125 (CA125) for ovarian and fallopian tube cancer and CA15.3 for breast cancer.
Results
Patients
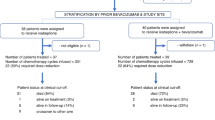
At time of data cutoff, 21 patients had been included. The first patient in this analysis was switched from combination treatment to olaparib monotherapy in June 2009, the last patient in May 2012. Baseline patient characteristics are presented in Table 1.
In total, 48% (10 out of 21) of all patients had breast cancer as the primary tumour type, 43% (9 out of 21) ovarian cancer and 10% (2 out of 21) had fallopian tube cancer. BRCA mutations were identified in 16 patients, of whom 81% (13 out of 16) had a BRCA1 mutation and 19% (3 out of 16) a BRCA2 mutation. Patients were heavily pre-treated, with 62% of patients having received three or more previous chemotherapy regimens. All patients had been previously treated with olaparib in the combination study. The reason for the switch was an adverse event in most cases (14 cases of persisting myelosuppression, 3 cases of peripheral neuropathy). Two patients were treated for localised disease after six cycles of combination therapy and continued on monotherapy and one patient requested the switch after eight cycles because she felt she could not tolerate the combination.
All of the patients had benefited from previous combination treatment, with eight patients having achieved a complete response (CR), eight partial response (PR) and five patients stable disease (SD).
Drug administration
Nearly all patients received olaparib monotherapy as capsules at 400 mg BID. One of the first patients who was switched to monotherapy received olaparib at 50 mg BID at first and was escalated to the known MTD of 400 mg BID in cycle 37 in order to achieve maximal exposure to olaparib. However, after 2 days this patient developed vasculitis on her legs that was also spreading to the feet. She also reported increased fatigue and gastrointestinal (GI) discomfort. For these reasons, a dose reduction of olaparib was applied at cycle 40–200 mg BID, which she tolerated well.
Safety
Treatment-related adverse events (TRAEs) observed during this study are summarised in Table 2. TRAEs were grouped per time period (cycle 1–6; cycle 7–12; cycle 13–24; cycle 25 and beyond) to explore whether a difference could be seen in the frequency and severity over time. The most common TRAE observed was bone marrow suppression as evidenced by neutropenia, anaemia, thrombocytopenia and lymphopenia. The incidence of these TRAEs was highest at baseline (43%, 14%, 33% and 10%, respectively) and over time the frequency of these events decreased (by cycle 25 and beyond the percentages were 20%, 0%, 20% and 0%, respectively). The severity of these events also decreased over time; although the first six cycles showed grade ⩾3 neutropenia (14%), anaemia, leuko- and lymphocytopenia (all 10%) and thrombocytopenia (5%), there was only one patient who showed a grade ⩾3 lymphocytopenia in later cycles. In addition to neutropenia, the only haematological adverse event that appeared to persist up to 24 cycles was anaemia (14% at baseline; 18% at cycle 13–24), for which some patients required frequent blood transfusions. Interestingly, in almost all patients this anaemia coincided with a significant increase in mean corpuscular volume (MCV). In fact, many patients showed MCV values above the normal range (80–100 fl) throughout olaparib treatment (see Figure 1), indicating that this anaemia was macrocytic. In one patient, a 54-year-old female with BRCA2-mutated breast cancer, a bone marrow analysis showed no abnormalities, in particular, no suspicion of myelodysplastic syndrome. Furthermore, laboratory values that could relate to the increase in MCV (e.g., folate, vitamin B12 and TSH) were measured, but no abnormalities were found.

MCV values before start of olaparib combination therapy and over time during treatment with olaparib. Median MCV values remained above the normal range during treatment with olaparib.
It is important to note that the haematological TRAEs did not lead to any clinically relevant toxicities such as neutropenic fever. All non-haematological TRAEs observed throughout this study were mild, only reaching grade 2 in three patients (fever, fatigue and rash). Most common non-haematological TRAEs at baseline were fatigue (38%), pain and nausea (both 19%) and cough, dyspnoea and diarrhoea (all 5%). Similar to the bone marrow suppression, the frequency of these TRAEs decreased over time (by cycle 13–24 the percentages were fatigue (9%), pain (18%), nausea, cough, dyspnoea and diarrhoea (all 0%)). Although the TRAEs were mild, some required concomitant medications (such as metoclopramide for nausea and loperamide for diarrhoea). Interestingly, the safety data and the need for use of concomitant proton pump inhibitors (PPIs) indicate that the incidence of GI toxicities such as oesophagitis, gastritis and dyspepsia increased with longer use of olaparib. In total, 11 out of 21 patients experienced GI complaints, mostly abdominal discomfort, with 7 of these patients having received 10 or more cycles. Concomitant treatment with PPIs reduced the complaints. In total, 49% of patients required a dose modification of olaparib. The dose modifications are presented per time period in Table 3. Most of these concerned dose interruptions. Reasons for dose modifications were TRAEs (mostly bone marrow suppression) or interruption of treatment for surgery or radiotherapy for localised recurrence of disease. Similar to the TRAEs, the requirement for dose modifications appeared to decrease over time; 62% (13 out of 21 patients) at baseline compared with 46% (5 out of 11 patients) in cycle 13–24. None of the patients included in this analysis had to omit a cycle or discontinue treatment due to TRAEs.
Anti-tumour activity
Figure 2 shows the treatment duration from the start of the combination study. Each bar represents one patient treated with olaparib, both the combination with chemotherapy and olaparib monotherapy. The patients are grouped by (1) mutation status and (2) tumour type. At time of data cutoff, all patients had undergone at least one disease evaluation. It is important to take into account baseline responses, as most of these patients had already shown anti-tumour response during the combination study. Best overall responses demonstrated nine (43%) CR, four (22%) PR, six (29%) SD, one (5%) progressive disease (PD) and one (5%) NE. Median treatment duration with olaparib monotherapy was 52 (7–183) weeks. Five (23%) patients experienced local recurrence during olaparib monotherapy for which they underwent surgery, radiotherapy or both, after which they were allowed to continue olaparib monotherapy. All of these patients showed continued benefit for months, although three patients eventually showed PD on CT scan and came off study.

Time on olaparib treatment. Patient are grouped by mutation status (BRCA1, BRCA2, wild-type or unknown). White bars indicate time on combination treatment; grey bars on monotherapy treatment. The columns indicate treatment for localised recurrence of disease and the black column indicates end of the treatment. Between brackets is the best overall response to olaparib treatment, whether in combination therapy or as monotherapy.
Responses stratified for mutation status are presented in Table 4. BRCA2 mutation carriers appeared to have the highest benefit with all three patients having CRs, compared with BRCA1 mutation carriers in whom there were six (46%) CRs and four (31%) PRs. In total 8 out of 16 (50%) patients with known BRCA mutations had to come off study due to PD. The median time on treatment was also longer for the BRCA2 vs BRCA1 mutation carriers (72 vs 52 weeks, respectively). Furthermore, median value of tumour markers appeared to slightly increase in BRCA1-mutated ovarian and fallopian tube carcinomas (CA125) over time, while these remained stable and within normal range in patients with BRCA2-mutated ovarian (CA125) and breast (CA15.3) cancer. In total, 9 out of 21 (43%) patients were still on the study at the time of data cutoff; 4 with breast, 3 with ovarian and 2 with fallopian tube cancer. When grouped by mutation status, 5 out of 13 (39%) patients with a BRCA1 mutation remained on study, three out of three (100%) patients with a BRCA2 mutation and one out of two (50%) patients with an unknown mutation status.
Discussion
Overall, the safety results show an encouraging long-term safety profile of olaparib. Incidences of the main TRAEs (mostly bone marrow suppression) that were most frequently seen at the switch to monotherapy and during the first six cycles decreased over time. This indicates that there was probably a carry-over effect of the chemotherapy. This hypothesis is strengthened by the recent publication of olaparib maintenance therapy trials in which the rates of myelosuppression were very low compared with this trial (Kaufman et al, 2014; Ledermann et al, 2014). Accordingly, the majority of patients (67%; 14 out of 21) had stopped the combination treatment and continued with olaparib monotherapy because of persisting neutro- and thrombocytopenia. The haematological TRAEs that persisted over time were neutropenia and anaemia. An interesting observation was that many patients experiencing recurring anaemia also showed an increased MCV. No mechanism has been described to date that could explain the role of olaparib in the development of macrocytic anaemia. This could hamper future double-blinded studies in which olaparib would be randomized against placebo, as MCV values could reveal which patient is receiving olaparib. The frequency of non-haematological TRAEs also decreased over time, again probably due to the carry-over effects of the chemotherapy used in the olaparib combination trial. Although the number of observations is small, the frequency of GI toxicities such as esophagitis, gastritis and dyspepsia appeared to increase after long-term treatment with olaparib, all easily manageable with PPIs. It might be argued that here may have been a selection bias over time, in that only the patients that tolerated the treatment well remained on study. However, none of the patients that went off study did so because of adverse events. Moreover, some patients had switched to olaparib monotherapy shortly before database lock and had thus only received olaparib monotherapy for 1–6 cycles. It is important to note that most of the patients who benefited from treatment and showed good tolerance remained on treatment well after database lock. Other serious toxicities that have previously been attributed to treatment with olaparib, such as pneumonitis or the development of secondary malignancies were not observed. However, a 71-year-old female patient with BRCA1-mutated ovarian cancer who had gone off study, went on to develop an aggressive form of non-Hodgkin lymphoma within 2 months after having received 10 cycles of olaparib monotherapy. The requirement for dose modifications also decreased over time, reflecting improving tolerability of olaparib monotherapy compared with combination treatment. Only four patients had to be dose reduced, two of whom within the first six treatment cycles, which was probably due to the carry-over effects of the chemotherapy. Interestingly, although one patient who was escalated from 50 to 400 mg BID did experience an increase in TRAEs, for which she was later dose reduced to 200 mg BID. Perhaps the most encouraging observation in this study was the long response duration after the switch to olaparib monotherapy. Nearly half of all patients were still on the study at the time of data cutoff, including four patients who had been on the study for >2 years. Interestingly, five patients demonstrated localised recurrence of disease during this study but continued on therapy after surgery and/or radiation therapy. Although this study provides us valuable information on safety and anti-tumour activity, it does come with several limitations. First of all, patients were all given olaparib as capsules, while in current trials olaparib is being administered as a tablet. However, although the tablets are known to give a higher exposure, the nature of the toxicities is not expected to differ between formulations. Second, additional data in the form of blood and/or tumour biopsies to collect pharmacokinetic and genetic biomarker data could have increased our understanding of the long-term effects of olaparib, as there were eight patients with BRCA mutations who did show disease progression. For example, in blood samples, it would have been interesting to see whether plasma olaparib levels remained stable over a prolonged period of time, as it has been reported that trough plasma concentrations of (for instance) imatinib decreased significantly after long-term treatment (Eechoute et al, 2012). Furthermore, olaparib doses might have increased after stopping paclitaxel, as data from the combination trial indicated a slight reduction in olaparib levels when co-administered with paclitaxel. This may have partly accounted for the persisting carry-over toxicities. In addition, biomarker data from tumour biopsies at the time of disease progression could have provided information on resistance mechanisms to olaparib in a patient population recognised as being sensitive to PARP inhibition. Finally, the durable responses were seen in a highly selected population (BRCA mutation carriers that had already responded to olaparib combination treatment), creating a form of selection bias. Also, the number of patients included in this analysis is relatively small (21 patients) when compared with other clinical trials, making it more difficult to draw significant conclusions on anti-tumour activity. Overall, although this study has provided encouraging evidence that treatment with olaparib monotherapy subsequent to being combined with chemotherapy is tolerable and effective in patients with BRCA-mutated tumours.
Change history
28 July 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ashworth A (2008) A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 26: 3785–3790.
Bennet N (2012) No survival benefit of olaparib in ovarian cancer patients. Lancet Oncol 13: 192.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917.
Bürkle A (2001) Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays 23: 795–806.
De Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Javier Oliver F, Masson M, Dierich A, LeMeur M, Waltzinger C, Chambon P, de Murcia G (1997) Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA 94: 7303–7307.
Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS (2011) Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell cycle 10: 1192–1199.
Eechoute K, Fransson MN, Reyners AK, de Jong FA, Sparreboom A, van der Graaf WTA, Friberg LE, Schiavon G, Wiemer EAC, Verweij J, Loos WJ, Mathijssen RHJ, De Giorgi U (2012) A long-term prospective population pharmacokinetic study on imatinib plasma concentrations in GIST patients. Clin Cancer Res 18: 5780–5787.
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NMB, Jackson SP, Smith GCM, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JHM, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumours from BRCA mutation carriers. N Engl J Med 361: 123–134.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A (2011) Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 12: 852–861.
Helleday T (2011) The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5: 387–393.
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, Bowen K, Fielding A, Domchek SM (2014) Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33: 244–250.
Khan OA, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, Carmichael J, Watson AJ, McGown G, Thorncroft M, Margison GP, Califano R, Larkin J, Wellman S, Middleton MR (2011) A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer 104: 750–755.
Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39: 8–24.
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin GJS, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Macpherson E, Dougherty B, Jürgensmeier JM, Orr M, Matulonis U (2013) Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer (SOC) and a BRCA mutation (BRCAm). J Clin Oncol (Meeting Abstracts) 31 (suppl): abstract 5505.
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin GJS, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U (2012) Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 366: 1382–1392.
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustni GJS, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, Barrett JC, Matulonis U (2014) Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15: 852–861.
Menear KA, Adcock C, Boulter R, Cockroft X, Copsey L, Cranston A, Dillon KL, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Matthews ITW, Moore S, O'Connor MJ, Smith GCM, Martin NMB (2008) 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem 51: 6581–6591.
Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RHJ, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, Mackay H, Rowbottom J, Lowe ES, Dougherty B, Barrett JC, Friedlander M (2015) Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol 16: 87–97.
Rajan A, Carter CA, Kelly RJ, Gutierrez M, Kummar S, Szabo E, Yancey MA, Ji J, Mannargudi B, Woo S, Spencer S, Figg WD, Giaccone G (2012) A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumours. Clin Cancer Res 18: 2344–2351.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AOH, Zander SAL, Derksen PWB, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O'Connor MJ, Martin NMB, Borst P, Jonkers J (2008) High sensitivity of BRCA1-deficient mammary tumours to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 105: 17079–17084.
Samol J, Ranson M, Scott E, Macpherson E, Carmichael J, Thomas A, Cassidy J (2012) Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumours: a phase I study. Invest New Drugs 30: 1493–1500.
Sandhu SK, Yap TA, de Bono JS (2010) Poly(ADP-ribose) polymerase inhibitors in cancer treatment: a clinical perspective. Eur J Cancer 46: 9–20.
Sodhi RK, Singh N, Jaggi AS (2010) Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul Pharmacol 53: 77–87.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumours. J Natl Cancer Inst 92: 205–216.
Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN, Rubin P (2003) CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol 13: 176–181.
Van der Noll R, Ang JE, Jager A, Marchetti S, Mergui-Roelvink M, de Bono JS, Lolkema MP, Brunetto A, Arkenau HT, de Jonge MJA, van der Biessen D, Tchakov I, Bowen K, Schellens JHM (2013a) Phase I study of olaparib in combination with carboplatin and/or paclitaxel in patients with advanced solid tumours. J Clin Oncol 31: abstract 2579.
Van der Noll R, de Grève J, Jager A, Marchetti S, Mergui-Roelvink M, de Jonge MJA, van der Biessen D, Tchakov I, Bowen K, Schellens JHM (2013b) Safety results from a Phase I study with a new tablet formulation of olaparib (O) in combination with carboplatin (C) and paclitaxel (Pa). Eur J Cancer 49: S174–S175.
Yap TA, Sandhu SK, Carden CP, de Bono JS (2011) Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J Clin 61: 31–49.
Acknowledgements
Dr JP de Boer, The Netherlands Cancer Institute, is gratefully acknowledged for performing the bone marrow biopsy and histological co-evaluation. The contribution of Y Groot, B Dufourny and P Hagen, Department of Clinical Pharmacology, The Netherlands Cancer Institute, Amsterdam, The Netherlands, to data monitoring and managing is gratefully acknowledged. The development team of olaparib led by Dr Ilian Tchakov of AstraZeneca is gratefully acknowledged for their support.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
van der Noll, R., Marchetti, S., Steeghs, N. et al. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer 113, 396–402 (2015). https://doi.org/10.1038/bjc.2015.256
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.256
Keywords
This article is cited by
-
Combined inhibition of BADSer99 phosphorylation and PARP ablates models of recurrent ovarian carcinoma
Communications Medicine (2022)
-
PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer
Clinical Pharmacokinetics (2018)
-
The effect of food on the pharmacokinetics of niraparib, a poly(ADP-ribose) polymerase (PARP) inhibitor, in patients with recurrent ovarian cancer
Cancer Chemotherapy and Pharmacology (2018)
-
Population pharmacokinetic analyses of the effect of carboplatin pretreatment on olaparib in recurrent or refractory women’s cancers
Cancer Chemotherapy and Pharmacology (2017)
-
Cytotoxic and targeted therapy for hereditary cancers
Hereditary Cancer in Clinical Practice (2016)
