
Abstract
Background:
The prognostic impact of segmental chromosome alterations (SCAs) in children older than 1 year, diagnosed with localised unresectable neuroblastoma (NB) without MYCN amplification enrolled in the European Unresectable Neuroblastoma (EUNB) protocol is still to be clarified, while, for other group of patients, the presence of SCAs is associated with poor prognosis.
Methods:
To understand the role of SCAs we performed multilocus/pangenomic analysis of 98 tumour samples from patients enrolled in the EUNB protocol.
Results:
Age at diagnosis was categorised into two groups using 18 months as the age cutoff. Significant difference in the presence of SCAs was seen in tumours of patients between 12 and 18 months and over 18 months of age at diagnosis, respectively (P=0.04). A significant correlation (P=0.03) was observed between number of SCAs per tumour and age. Event-free (EFS) and overall survival (OS) were calculated in both age groups, according to both the presence and number of SCAs. In older patients, a poorer survival was associated with the presence of SCAs (EFS=46% vs 75%, P=0.023; OS=66.8% vs 100%, P=0.003). Moreover, OS of older patients inversely correlated with number of SCAs (P=0.002). Finally, SCAs provided additional prognostic information beyond histoprognosis, as their presence was associated with poorer OS in patients over 18 months with unfavourable International Neuroblastoma Pathology Classification (INPC) histopathology (P=0.018).
Conclusions:
The presence of SCAs is a negative prognostic marker that impairs outcome of patients over the age of 18 months with localised unresectable NB without MYCN amplification, especially when more than one SCA is present. Moreover, in older patients with unfavourable INPC tumour histoprognosis, the presence of SCAs significantly affects OS.
Similar content being viewed by others

Main
Peripheral neuroblastic tumours are a heterogeneous group of cancers with regard to clinical, genetic and histopathological classification, the most frequent being neuroblastoma (NB). Stage and age at diagnosis are important prognostic clinical features (Breslow and McCann, 1971; London et al, 2005a, 2005b; Deyell and Attiyeh, 2011), but disease outcome is also strongly influenced by the presence of genomic alterations in neuroblastic cells, for example, amplification of the MYCN oncogene, a powerful prognostic marker that occurs in about 20% of the patients (Brodeur et al, 1984; Seeger et al, 1985; Sansone et al, 1991; Tonini et al, 1997, 2012).
Segmental chromosome alterations (SCAs) other than MYCN amplification have been described as associated with tumour aggressiveness (Janoueix-Lerosey et al, 2009; Schleiermacher et al, 2010; Coco et al, 2012). Janoueix-Lerosey et al (2010) suggested a risk criteria assessment based on five classes of genomic alterations, ranging from tumours with only numerical alterations and the absence of disease-related deaths, to tumours with MYCN amplification and SCAs in patients with a poor outcome (Ambros et al, 2009; Janoueix-Lerosey et al, 2009). On the basis of these observations, the ‘multilocus’ (i.e., Multiplex Ligation Probe Amplification) or ‘pangenomic’ (i.e., array-based) approach to define tumour genomic profile for therapeutic stratification is a potential important tool to guide therapy (Janoueix-Lerosey et al, 2009).
In 2001, the SIOPEN (International Society of Paediatric Oncology European Neuroblastoma) launched the European Unresectable Neuroblastoma (EUNB) study for the treatment of children over 1 year of age with localised unresectable NB, defined by the presence of surgical risk factors (SRFs) and without MYCN amplification. Clinical results of the study have already been published elsewhere (Kohler et al, 2013). After early biopsy, chemotherapy was the initial approach for this group of patients followed, when possible, by delayed surgery. As these patients did not undergo tumour resection at diagnosis, an accurate characterisation of the biological features of the tumour to detect genetic alterations of prognostic significance following tumour biopsy, was of the upmost importance. On the basis of these considerations, we have analysed tumours from these patients to detect the presence of SCAs and their potential influence on survival in this uniformly treated cohort.
Patients and methods
Patients
Between January 2001 and October 2006, 160 patients from 10 different European countries (Austria, Belgium, Czech Republic, France, Italy, Norway, Portugal, Spain, Sweden, and United Kingdom) were enrolled in the EUNB protocol with a new diagnosis of localised unresectable NB, as defined by the presence of SRFs. Informed consent was obtained from patients’ parents or guardians. A pangenomic/multilocus approach was possible for tumour samples from 98 patients.
Tumour samples
Tumours samples at diagnosis were centrally collected at the national reference centres and processed according to the standard procedures of the SIOPEN Biology European Neuroblastoma Quality Assessment (ENQUA) Group (Ambros et al, 2001). Only samples with ⩾60% of neuroblastic cells were considered for array CGH and Multiplex Ligation Probe Amplification (MLPA) analysis.
Multiplex Ligation Probe Amplification
The MLPA (SALSA MLPA kit p251/A1-B1, p252/A1-B1, p253/A1-B1 neuroblastoma, MRC Holland, Amsterdam, The Netherlands) was performed on DNA samples as previously described (Ambros et al, 2011). The neuroblastoma MLPA kits are designed to assess the status of selected regions in the following chromosomes: 1, 2, 3, 4, 7, 9, 11, 12, 14, and 17.
Array CGH
Following standardised extraction, 30 DNA samples were analysed by array CGH using an in-house BAC/PAC array with a genomic resolution of ∼1 Mb, as reported previously (Janoueix-Lerosey et al, 2009, 2010) and 9 by a commercially available NimbleGen DNA array (Roche NimbleGen, Madison, WI, USA) containing 72 000 oligonucleotide probes with an average resolution of ∼1 probe per 40 kb.
Nomenclature of SCAs
The presence and number of SCAs frequently occurring in NB (see Results) were classified according to the SIOPEN Biology guidelines (Ambros et al, 2011). No SCA is indicated by the code s0, where ‘s’ stands for ‘segmental’. In the cases of SCAs, the code indicates the number of SCAs (s1, s2, s3, and so on). The presence of an unbalanced ratio between the signals of chromosomal regions of interest (decreased/increased mean value of more than 0.25 of at least two adjacent probes as compared with the mean values of the remaining probes of the concerned chromosome/chromosomal arm) and the reference signals defined a SCA by MLPA (Ambros et al, 2011). Segmental chromosome alterations identified by array CGH were defined by the presence of either at least 3 adjoining BAC/PAC clones or 100 contiguous oligonucleotide probes showing a genomic status different from that of the rest of the chromosome (Schleiermacher et al, 2011).
Statistical analysis
Age at diagnosis was categorised into two groups using 18 months as age cutoff. Disease progression, relapse, and death were considered as events. Event-free (EFS) and overall survival (OS) were estimated by the Kaplan–Meier method and differences between groups were assessed by the log-rank test. Survival estimates referred to 5 years from diagnosis and the related 95% confidence intervals (95% CIs) were calculated by the Kalbfleisch and Prentice method (Kalbfleisch and Prentice, 1980). All statistical tests were two-sided and a P-value of <0.05 was considered as significant. All analyses were performed using the statistical package ‘Stata’ (release 11.0, Stata Corporation, College Station, TX, USA).
Results
A multilocus/pangenomic approach was possible for 98 out of 160 (61.2%) patients enrolled in the EUNB study. For the remaining 62 patients, there was insufficient tumour available for genetic studies or samples contained a low percentage of neoplastic cells. Of the 98 patients, 38 (38.8%) were 12–18 months of age at diagnosis and 60 (61.2%) were older than 18 months. Centrally reviewed INPC (International Neuroblastoma Pathology Classification) classification assessed by the pathology panel was available for 68 out of 98 patients (69.4%), of whom 28 (21 were 12–18 months and 7 over 18 months at diagnosis) had favourable and 40 (5 were 12–18 months and 35 over 18 months at diagnosis) unfavourable histoprognosis. To exclude any selection bias, the 98 patients were compared with the 62 not studied by multilocus/pangenomic approach, with regard to age, sex, stage, country of origin, tumour site, type of biopsy, relapses, deaths, and INPC (Shimada et al, 1999). No significant differences were observed between the two groups (Supplementary Table 1; Supplementary Figure 1).
The MLPA and array CGH techniques were used to study 59 and 39 tumour samples, respectively. High intertechnique concordance has been previously described by Ambros et al (2011). All data were centrally reviewed by the ENQUA Biology group. The current study is a separate cohort from that previously studied by Ambros et al (2011) and Schleirmacher et al (2011).
Since MLPA gave results on the status of selected genetic loci of specific chromosomes while array CGH allowed the evaluation of all the chromosomes, combined results of the two techniques were reported only for those regions included in the MLPA kit. Forty-four tumours (44.9%) did not show any SCA (s0). In 54 tumours (55.1%) the multilocus/pangenomic approach showed at least 1 SCA out of the 7 mostly recurrent in NB, namely gain of 1q, 2p, 17q and loss of 1p, 3p, 4p, 11q. The following SCAs, listed by frequency, were observed: 17q gain (35%; 34 out of 97), 1p loss/1q gain (26%; 24 out of 93), 11q loss (25%; 24 out of 96), 2p gain (17.5%; 17 out of 97), 3p loss (7%; 7 out of 96), and 4p loss (4%; 4 out of 97). Among the other chromosomes included in the MLPA kit, we detected gain of 7q in 10% of samples (10 out of 98), 12q gain in 4% (4 out of 98), and 14q loss in 3% (3 out of 97). Among the chromosomes analysed by aCGH only, a deletion of 6q was seen in 10% of the tumour samples (4 out of 39). With respect to the number of SCAs, 21 tumours (38.9%) were classified as s1, 17 (31.5%) as s2, 10 (18.5%) as s3, and 6 (11.1%) as s4.
Henceforth when reporting information on SCAs, we will refer only to the seven recurrent SCAs in NB reported above, detected either by MLPA or by array CGH.
Since age at diagnosis has an important role in patient risk assessment, as reported by Kohler et al (2013), we stratified patients by 18 months age cutoff. The percentage of tumours with at least 1 of the 7 recurrent SCAs in NB was significantly greater in the 60 patients older than 18 months of age compared with the 38 ones 12–18 months old (63% vs 42%; P=0.04).
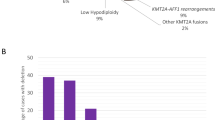
Moreover, the number of SCAs per tumours (i.e., no SCA, 1 SCA or more than 1 SCA) significantly correlated with age at diagnosis, as only 1 of 38 patients aged less than 18 months (2.6%) had more than two SCAs, while 15 of 60 older patients (25%) showed more than two SCAs (P=0.031) (Figure 1).

Distribution of the number of SCAs in tumours of patients under and over 18 months of age at diagnosis. On the left side the number of tumours without (s0) or with (s1, s2, s3, s4) SCAs in patients less than 18 months at diagnosis, on the right side the number of tumours without (s0) or with (s1, s2, s3, s4) SCAs in patients over 18 months. The graph shows that tumours of older patients have increased number of SCAs, in particular three or more SCAs per tumour.
Overall and event-free survival were calculated for both age groups according to the presence of SCAs. For children aged 12–18 months, the presence of SCAs did not influence survival (OS 100% vs 100%, EFS 100% vs 95.5%, P=0.45). In particular, 22 patients with tumours without SCAs experienced two events and only one event occurred in the 16 patients with tumours with SCAs (data not shown).
For children older than 18 months at diagnosis, the presence of SCAs was significantly associated with OS (100% children without SCAs vs 66.8% children ⩾1 SCA, P=0.003) (Figure 2A) and with EFS (75% vs 46.1%, P=0.023) (Figure 2B).

Kaplan–Meier survival analysis showing 5-year OS (A) and EFS (B) in patients over than 18 months of age at diagnosis according to the presence SCAs. Presence of SCAs refers to the seven most common SCAs observed in neuroblastoma (1p−, +1q, +2p, 3p−, 4p, 11q−, +17q). (A) OS was 100% in the absence of SCA and 66.8% in the presence of SCAs; (B) EFS was 75% in the absence of SCAs and 46.1% in the presence of SCAs.
The prognostic significance of each of the seven recurrent SCAs with respect to both OS and EFS has been evaluated before and after age stratification. Results for the whole cohort and for children over 18 months of age are reported in Table 1. There was a significant correlation between all the SCAs, but for 3p loss, and OS, without any differences according to age stratification, except for 11q loss (Table 1). Event-free survival did correlate with 4p loss, 11q loss and 17q gain in the whole cohort, without any differences according to age stratification. Results for the group 12–18 months were not statistically significant (Supplementary Table 2).
We further calculated OS and EFS according to the number of SCAs in the two groups based on the age at diagnosis. There was no significant correlation between EFS and number of SCAs in both groups (Supplementary Figure 2). Overall survival was significantly associated with the number of SCAs in patients over 18 months of age (OS 100% vs 84.6% vs 57.9%, P=0.002) (Figure 3). We also studied the impact of SCAs in patients with both favourable and unfavourable histoprognosis. A statistically significant association between a poor OS and the presence of SCAs (OS 100% vs 61%, P=0.0183) (Figure 4A) was observed in patients over 18 months with unfavourable histoprognosis, while EFS was not influenced (Figure 4B).

Kaplan–Meier survival analysis showing 5-year OS according to the presence of one or more SCAs in tumours of patients under (A) and over (B) 18 months of age at diagnosis. The figure shows survival curves of patients with tumour harbouring no SCA, one and more than one SCAs (see also text). (A) OS was 100% irrespective to the number of SCAs; (B) OS was 100% in patients without SCAs, 84.6% in patients with one SCA and 57.9% in patients with two or more SCAs.

Kaplan–Meier survival analysis showing 5-year OS (A) and EFS (B) according to the presence of SCAs in patients over 18 months at diagnosis and unfavourable histoprognosis. (A) OS was 100% in the absence of SCAs and 61% in the presence of SCAs; (B) EFS was 65.9% in the absence of SCAs and 45.5% in the presence of SCAs.
Discussion
Patients enrolled in the EUNB protocol represent a cohort of patients whose tumours have normal MYCN gene status, suitable to be investigated for the presence of SCAs and their role in disease outcome. In this study, the genomic pattern of the tumour cells was analysed by multilocus/pangenomic assays with the aim to identify possible correlations between chromosome abnormalities other than MYCN amplification and the occurrence of adverse disease events.
MYCN gene amplification, described in about 20% of NB, is one of the most reliable prognostic markers and is used to determine treatment in infants and in children with localised disease. In patients whose tumour lacks MYCN amplification, the poor prognosis could be associated with the presence of other SCAs. This indicates that SCAs have an important role in disease progression and their presence is a useful prognostic marker in NB.
Age at diagnosis was categorised into two groups by 18 months as age cutoff (12–18 and more than 18 months) to verify whether the SCAs correlated with age. The presence of SCAs differed significantly in the two patient groups and, in addition, tumours of older children had a significantly higher number of SCAs compared with those of the younger ones, confirming previous results (Schleiermacher et al, 2010; Coco et al, 2012; Stigliani et al, 2012). Regarding the impact of SCAs on disease course, we observed a significant correlation between poor prognosis in older children and both the presence and number of SCAs, irrespective of the chromosome involved. This observation indicates that the presence of any SCA, in particular more than one, rather than a specific SCA, affects the disease course. Essentially, in the cohort of 98 patients, as well as in the older subgroup, all types of SCAs, except 3p loss, were associated with a poor OS and in the older group, statistical significance for 11q loss was borderline. Chromosome 11q loss was significantly associated with worse OS and EFS (both P=0.035) in the whole cohort of patients. Loss of 11q, a negative prognostic factor that is usually inversely associated with MYCN amplification (Fisher et al, 2010), was detected in 25% of cases, mostly in patients over 18 months of age at diagnosis (namely, 7 out of 38 under and 17 out of 60 over 18 months, respectively). It has been suggested that 11q loss is associated with a poor prognosis in older children (Carén et al, 2010). Our results indicate that neither MYCN amplification nor 11q deletion are the unique chromosome defects associated with NB progression but rather accumulation of diverse SCAs have a critical impact in tumour progression.
Taking into account the results of array CGH alone, we observed 6q loss in 4 out of 39 (10%) patients, 3 of whom were older than 18 months. Our observation is in accordance with previous results from Stallings et al (2006), who reported 8% of 6q loss in a subset of 49 NB. Michels et al (2007) showed 6q loss in 55% of NB cell lines and in 2 out of 75 (3%) primary tumours. Although chromosome 6 is frequently involved in other malignancies, (Mitelman et al) 6q loss is a rare event in NB (Michels et al, 2007). Patients older than 18 months of age, whose tumour shows 6q loss, had significant poor EFS (P=0.046, data not shown) compared with those without, but we think that the number of cases is too small to draw any conclusion.
Data reported by Kohler et al (2013) regarding the clinical course of patients enrolled in the EUNB study showed that the group of patients with unfavourable tumour histoprognosis and older age had a worse outcome, suggesting that more intensive treatment for this group of patients is needed. On the basis of these data, we studied the potential significance of the presence of SCAs in the subgroup of patients with INPC unfavourable tumour histoprognosis. Of note, a statistically significant correlation between a poor OS and the presence of SCAs was observed in patients over 18 months. Event-free survival was not influenced by SCAs, as a similar number of events occurred in patients of both groups of age.
Overall, our data show that both the presence and number of SCAs are associated with older age and represent a poor prognostic marker for patients over 18 months of age at diagnosis, with localised unresectable MYCN non-amplified NB. Moreover, older patients with INPC unfavourable tumour histoprognosis, whose tumours do not present SCAs, are more likely to survive after relapse than patients with tumour with SCAs. Future clinical trials of this group of patients should be stratified by the presence and number of SCAs.
In conclusion, this study shows an example of tumour molecular analysis in an homogenous group of unresectable NB patients enrolled in a unique clinical trial. Although it was not possible to collect tumour samples from all patients, the homogeneity of the records made up for this lack. Our study also indicates that close cooperation among oncologists, surgeries, pathologists, and biologists is mandatory to recover as many biological samples as possible and to give to the patient the best diagnosis and treatment. In the future, this close cooperation should be strengthened, together with a more standardisation of the molecular approaches.
Change history
20 January 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ambros IM, Brunner B, Aigner G, Bedwell C, Beiske K, Bénard J, Bown N, Combaret V, Couturier J, Defferrari R, Gross N, Jeison M, Lunec J, Marques B, Martinsson T, Mazzocco K, Noguera R, Schleiermacher G, Speleman F, Stallings R, Tonini GP, Tweddle DA, Valent A, Vicha A, Roy NV, Villamon E, Ziegler A, Preuner S, Drobics M, Ladenstein R, Amann G, Schuit RJ, Pötschger U, Ambros PF (2011) A multilocus technique for risk evaluation of patients with neuroblastoma. Clin Cancer Res 17 (4): 792–804.
Ambros PF, Ambros IM SIOP Europe Neuroblastoma Pathology, Biology and Bone Marrow Group (2001) Pathology and biology guidelines for resectable and unresectable neuroblastic tumors and bone marrow examination guidelines. Med Pediatr Oncol 37 (6): 492–504.
Ambros PF, Ambros IM, Brodeur GM, Haber M, Khan J, Nakagawara A, Schleiermacher G, Speleman F, Spitz R, London WB, Cohn SL, Pearson AD, Maris JM (2009) International consensus for neuroblastoma molecular diagnostic: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100 (9): 1471–1482.
Breslow N, McCann B (1971) Statistical estimation of prognosis for children with neuroblastoma. Cancer Res 31 (12): 2098–2103.
Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM (1984) Amplification of N-MYC in untreated human neuroblastomas correlates with advanced disease stage. Science 224 (4653): 1121–1124.
Carén H, Kryh H, Nethander M, Sjöberg RM, Träger C, Nilsson S, Abrahamsson J, Kogner P, Martinsson T (2010) High-risk neuroblastoma tumours with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci USA 107 (9): 4323–4328.
Coco S, Theissen J, Scaruffi P, Stigliani S, Moretti S, Oberthuer A, Valdora F, Fischer M, Gallo F, Hero B, Bonassi S, Berthold F, Tonini GP (2012) Age-dependent accumulation of genomic aberrations and deregulation of cell cycle and telomerase genes in metastatic neuroblastoma. Int J Cancer 131 (7): 1591–1600.
Deyell RJ, Attiyeh EF (2011) Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer Genet 204 (3): 113–121.
Fisher M, Bauer T, Oberthur A, Hero B, Theissen J, Ehrich M, Spitz R, Eils R, Westermann F, Brors B, König R, Berthold F (2010) Integrated genomic profiling identifies two distinct molecular subtypes with divergent outcome in neuroblastoma with loss of chromosome 11. Oncogene 29 (6): 865–875.
Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, Vermeulen J, Couturier J, Peuchmaur M, Valent A, Plantaz D, Rubie H, Valteau-Couanet D, Thomas C, Combaret V, Rousseau R, Eggert A, Michon J, Speleman F, Delattre O (2009) Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27 (7): 1026–1033.
Janoueix-Lerosey I, Schleiermacher G, Delattre O (2010) Molecular pathogenesis of peripheral neuroblastic tumours. Oncogene 29 (11): 1566–1579.
Kalbfleisch JD, Prentice RL (1980) The Statistical Analysis of Failure Time Data. John Wiley & Sons: New York.
Kohler JA, Rubie H, Castel V, Beiske K, Holmes K, Gambini C, Casale F, Munzer C, Erminio G, Parodi S, Navarro S, Marquez C, Peuchmaur M, Cullinane C, Brock P, Valteau-Couanet D, Garaventa A, Haupt R (2013) Treatment of children over the age of one year with unresectable localised neuroblastoma without MYCN amplification: results of the SIOPEN study. Eur J Cancer 49 (17): 3671–3679.
London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP, Cohn SL (2005a) Evidence for an age cut off greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol 23 (27): 6459–6465.
London WB, Boni L, Simon T, Berthold F, Twist C, Schmidt ML, Castleberry RP, Matthay KK, Cohn SL, De Bernardi B (2005b) The role of age in neuroblastoma risk stratification: the German, Italian and Children’s Oncology Group perspectives. Cancer Lett 228 (1-2): 257–266.
Michels E, Vandesompele J, De Preter K, Hoebeeck J, Vermeulen J, Schramm A, Molenaar JJ, Menten B, Marques B, Stallings RL, Combaret V, Devalck C, De Paepe A, Versteeg R, Eggert A, Laureys G, Van Roy N, Speleman F (2007) ArrayCGH-based classification of neuroblastoma into genomic subgroups. Genes Chromosomes Cancer 46 (12): 1098–1108.
Mitelman F, Johansson B, Mertens F Mitelman Database of Chromosome Aberrations in Cancer. Available at http://cgap.nci.nih.gov/Chromosome/Mitelman.
Sansone R, Strigini P, Badiali M, Dominici C, Fontana V, Iolascon A, De Bernardi B, Tonini GP (1991) Age-dependent prognostic significance of N-myc amplification in neuroblastoma. The Italian experience. Cancer Genet Cytogenet 54 (2): 253–257.
Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, Klijanienko J, Couturier J, Pierron G, Mosseri V, Valent A, Auger N, Plantaz D, Rubie H, Valteau-Couanet D, Bourdeaut F, Combaret V, Bergeron C, Michon J, Delattre O (2010) Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28 (19): 3122–3130.
Schleiermacher G, Michon J, Ribeiro A, Pierron G, Mosseri V, Rubie H, Munzer C, Bénard J, Auger N, Combaret V, Janoueix-Lerosey I, Pearson A, Tweddle DA, Bown N, Gerrard M, Wheeler K, Noguera R, Villamon E, Cañete A, Castel V, Marques B, de Lacerda A, Tonini GP, Mazzocco K, Defferrari R, De Bernardi B, di Cataldo A, van Roy N, Brichard B, Ladenstein R, Ambros I, Ambros P, Beiske K, Delattre O, Couturier J (2011) Segmental chromosomal alterations lead to a high risk of relapse in infants with MYCN-non amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105 (12): 1940–1948.
Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D (1985) Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 313 (18): 1111–1116.
Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B (1999) Terminology and morphologic criteria of neuroblastic tumours: recommendations by the International Neuroblastoma Pathology Committee. Cancer 86 (2): 349–363.
Stallings RL, Nair P, Maris JM, Catchpoole D, McDermott M, O'Meara A, Breatnach F (2006) High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res 66 (7): 3673–3680.
Stigliani S, Coco S, Moretti S, Oberthuer A, Fischer M, Theissen J, Gallo F, Garaventa A, Berthold F, Bonassi S, Tonini GP, Scaruffi P (2012) High genomic instability predicts survival in metastatic high-risk neuroblastoma. Neoplasia 2012 14 (9): 823–832.
Tonini GP, Boni L, Pession A, Rogers D, Iolascon A, Basso G, Cordero di Montezemolo L, Casale F, Pession A, Perri P, Mazzocco K, Scaruffi P, Lo Cunsolo C, Marchese N, Milanaccio C, Conte M, Bruzzi P, De Bernardi B (1997) MYCN oncogene amplification in neuroblastoma is associated with worse prognosis, except in stage 4s: the Italian experience with 295 children. J Clin Oncol 15 (1): 85–93.
Tonini GP, Nakagawara A, Berthold F (2012) Towards a turning point of neuroblastoma therapy. Cancer Lett 326 (2): 128–134.
Acknowledgements
This work was supported by: Italian Neuroblastoma Foundation; Cancer Research UK (CRUK) and the Neuroblastoma Society in the UK; MH CZ-DRO; University Hospital Motol, Prague, Czech Republic 00064203; grants from the Fundación Asociación Española contra el Cáncer, FIS (contract PI10/15) and RTICC (contracts RD06/0020/0102; RD12/0036/0020), Instituto Carlos III Madrid & ERDF, Spain; the National Resource Centre for Childhood Solid Tumours (KSSB), Norway; grants from the Institut National de la Santé et de la Recherche Médicale, the Ligue Nationale Contre le Cancer (Equipe labellisée) the Federation Enfants et Sante and the Societe Francaise de Lutte contre les Cancers et les Leucemie de l’Enfant et de l’Adolescent (SFCE) and the PHRC AOM 02014. The construction of the BAC/PAC array was supported by grants from the Carte d’Identité des Tumeurs programme of the Ligue Nationale Contre le Cancer. RD and EG are supported by Italian Neuroblastoma Foundation. GS is supported by the Annenberg Foundation. The work of IMA and PFA was supported by the Austrian National Bank, grant number: 13422 and the CCRI. We thank the associations APAESIC (Association des Parents et des Amis des Enfants Soignés à l’Institut Curie), Association Hubert Gouin, ‘Les Bagouz à Manon’ and ‘Enfants et Santé’. We thank Dr Bruno De Bernardi for helpful suggestions in preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Defferrari, R., Mazzocco, K., Ambros, I. et al. Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112, 290–295 (2015). https://doi.org/10.1038/bjc.2014.557
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.557
Keywords
This article is cited by
-
Mutations of 1p genes do not consistently abrogate tumor suppressor functions in 1p-intact neuroblastoma
BMC Cancer (2022)
-
Distribution of segmental chromosomal alterations in neuroblastoma
Clinical and Translational Oncology (2021)
-
Systematic computational identification of prognostic cytogenetic markers in neuroblastoma
BMC Medical Genomics (2019)
-
Ganglioneuroblastoma in children
Neurological Sciences (2019)
-
Neuroblastoma: clinical and biological approach to risk stratification and treatment
Cell and Tissue Research (2018)
