
Abstract
Background:
This phase II, open-label, randomised study evaluated whether patients with metastatic pancreatic cancer receiving erlotinib/gemcitabine derived survival benefits from increasing the erlotinib dose.
Methods:
After a 4-week run-in period (gemcitabine 1000 mg m−2 once weekly plus erlotinib 100 mg per day), patients with metastatic pancreatic cancer who developed grade 0/1 rash were randomised to receive gemcitabine plus erlotinib dose escalation (150 mg, increasing by 50 mg every 2 weeks (maximum 250 mg); n=71) or gemcitabine plus standard-dose erlotinib (100 mg per day; n=75). The primary end point was to determine whether overall survival (OS) was improved by increasing the erlotinib dose. Secondary end points included progression-free survival (PFS), incidence of grade ⩾2 rash, and safety.
Results:
Erlotinib dose escalation induced grade ⩾2 rash in 29 out of 71 (41.4%) patients compared with 7 out of 75 (9.3%) patients on standard dose. Efficacy was not significantly different in the dose-escalation arm compared with the standard-dose arm (OS: median 7.0 vs 8.4 months, respectively, hazard ratio (HR), 1.26, 95% confidence interval (CI): 0.88–1.80; P=0.2026; PFS: median 3.5 vs 4.5 months, respectively, HR, 1.09, 95% CI: 0.77–1.54; P=0.6298). Incidence of adverse events was comparable between randomised arms.
Conclusion:
The erlotinib dose-escalation strategy induced rash in some patients; there was no evidence that the higher dose translated into increased benefit.
Similar content being viewed by others

Main
Treatment options for pancreatic cancer remain limited and until 2005, single-agent gemcitabine was the standard of care. Fluorouracil (5-FU), capecitabine, cisplatin, docetaxel, irinotecan, oxaliplatin, pemetrexed, and cetuximab additions showed no clear survival benefits, and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and 5-FU) had high toxicity (Philip et al, 2007; Burris and Rocha-Lima, 2008; Merl et al, 2010; Conroy et al, 2011). A better understanding of pancreatic cancer and the role of epidermal growth factor receptor (EGFR) overexpression in cell proliferation/metastasis (Tobita et al, 2003) led to molecularly-targeted agents. In the National Cancer Institute of Canada Clinical Trials Group phase III PA.3 study of advanced pancreatic cancer, erlotinib, an EGFR tyrosine-kinase inhibitor (TKI), plus gemcitabine, increased overall survival (OS; hazard ratio (HR), 0.82; 95% confidence interval (CI): 0.69–0.99; P=0.038; median 6.2 vs 5.9 months, respectively) and 1-year survival rate (23% vs 17%, respectively; P=0.023) vs single-agent gemcitabine (Moore et al, 2007). Erlotinib plus gemcitabine was approved for the first-line treatment of advanced pancreatic cancer.
In PA.3, the clinical benefit of erlotinib was significant, but modest; the use of biomarkers to individualise EGFR TKI therapy may optimise treatment, as seen with EGFR mutation-positive non-small-cell lung cancer (NSCLC) (Zhou et al, 2011; Rosell et al, 2012). No specific biomarker has been identified for erlotinib-treated pancreatic cancer (da Cunha Santos et al, 2010); however, there is evidence that rash could be a surrogate marker of efficacy (Clark et al, 2003; Saltz et al, 2003; Shepherd et al, 2005; Wacker et al, 2007; Verslype et al, 2009; Van Cutsem et al, 2009a; Buges et al, 2012; Perez-Soler et al, 2012). Correlations with rash have also been noted with the anti-EGFR agents cetuximab (Van Cutsem et al, 2009b) and panitumumab (Peeters et al, 2009). A dose-response relationship or differences in gene expression and immune response that simultaneously affect rash development and tumour response may underpin this (Amador et al, 2004; Guttman-Yassky et al, 2010). Systematically increasing the dose to induce rash has shown promise in colorectal cancer; the EVEREST study demonstrated a higher overall response rate (ORR) when cetuximab was 'dosed to rash' compared with standard dosing (Van Cutsem et al, 2012).
We describe a phase II study to determine whether dose escalation of erlotinib translated into improved clinical outcomes in patients with advanced pancreatic cancer.
Materials and methods
Study design
RACHEL (BO21128; ClinicalTrials.gov: NCT00652366; http://clinicaltrials.gov/ct2/show/NCT00652366) was a multicentre, randomised, open-label, phase II study investigating gemcitabine plus standard-dose or escalated-dose erlotinib in patients with metastatic pancreatic cancer (Figure 1).

Study design of the phase II RACHEL (BO21128) study. The 4-week run-in period was based on a median onset for rash development in patients treated with erlotinib plus gemcitabine for 10 days (with 90% of patients experiencing rash within 44 days) seen in the PA.3 study (Wacker et al, 2007). Erlotinib dose escalation—starting at 150 mg per day increasing at 50 mg every 2 weeks until a maximum of 250 mg per day or grade ⩾2 rash developed (patients were held at the lowest dose that induced grade ⩾2 rash). A 50-mg increment allowed monitoring of rash development/other toxicities and ensured patients were not exposed to unnecessarily high doses of erlotinib. Abbreviations: ECOG PS=Eastern Cooperative Oncology Group performance status; OS=overall survival; PD=disease progression; PFS=progression-free survival.
The RACHEL trial was approved by the institutional review board/ethics committee of each centre, and was performed in accordance with the principles of the Declaration of Helsinki and Guidelines for Good Clinical Practice. Patients provided written informed consent.
Patients
Patients (⩾18 years) had: histologically/cytologically confirmed pancreatic cancer (adenocarcinoma) with measurable/non-measurable disease (stage IV); Eastern Cooperative Oncology Group performance status (ECOG PS) 0 to 1; life expectancy ⩾8 weeks; and adequate haematological/renal/liver function. Eligible patients completed a 4-week run-in period. The inclusion criteria for randomisation were completion of the run-in period: without evidence of disease progression (PD); without grade ⩾2 rash development; or without development of toxicity leading to dose adjustments <75% of the full chemotherapy dose. The main exclusion criteria were: local (stage Ia to IIb) pancreatic cancer and locally advanced (stage III) pancreatic cancer; prior chemotherapy or treatment with another systemic anti-cancer agent for locally advanced unresectable/metastatic pancreatic cancer; ⩽6 months since last adjuvant therapy; prior EGFR TKIs treatment or adjuvant radiotherapy; any other malignancies within the last 5 years (except adequately treated carcinoma in situ of the cervix or basal/squamous cell skin cancer); spinal cord compression; and central nervous system metastases.
Assessments
Tumour response was evaluated according to the Response Evaluation Criteria in Solid Tumors (RECIST; version 1.0) 2 weeks (maximum) before the first dose, and then at weeks 8, 16, 24, 32, and every 12 weeks thereafter until PD. A formalin-fixed, paraffin-embedded pancreatic tumour block or slides were required (if available). Mandatory serum samples for biomarkers were taken before treatment, in cycle 3, and at PD. Adverse events (AEs), including rash, were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAEs; version 3.0).
Study treatment
During the run-in period, patients were treated with weekly gemcitabine 1000 mg m−2 (intravenously (IV)) plus daily erlotinib 100 mg per day (oral) for 4 weeks. After the run-in period, patients with no rash (grade 0) or grade 1 rash were randomised to the standard-dose or dose-escalation arms (central randomisation: interactive voice/web response system). The study sponsor was blinded to treatment allocation. Patients were initially considered non-eligible for randomisation if they did not receive the full run-in period of both gemcitabine and erlotinib. As many patients could not receive the full gemcitabine dose, the protocol was amended to allow dose reductions to ⩾75%; patients with only one gemcitabine-related dose reduction for haematological toxicity were eligible, provided they were stable and tolerating the 75% dose and no further reductions were anticipated. Patients on the standard-dose arm received gemcitabine 1000 mg m−2 (IV; weekly for 3 consecutive weeks, followed by a 1-week treatment holiday) plus daily oral erlotinib 100 mg per day until PD, unacceptable toxicity, death, or patient withdrawal. Patients on the dose-escalation arm received gemcitabine (as above) plus daily oral erlotinib (150 mg per day, increased in 50-mg increments bi-weekly until development of grade ⩾2 rash (maximum 250 mg) or other dose-limiting toxicity) until unacceptable toxicity/PD, death, or study withdrawal. Patients with grade 0 or 1 rash after the run-in period (who were non-eligible for randomisation) or patients who had grade ⩾2 rash after the run-in period, continued to receive the standard dose of erlotinib and gemcitabine (as above; non-randomised arm). Dose reductions (50 mg per day decrements) for AEs were allowed. Patients who progressed during the run-in period were discontinued from the study.
Efficacy and safety analyses
The primary objective was to determine whether OS was improved by erlotinib dose escalation to induce rash in patients who developed grade 0 or 1 rash during a 4-week run-in period with standard-dose erlotinib plus gemcitabine, compared with patients who continued to receive standard-dose erlotinib plus gemcitabine. The secondary objectives were: to evaluate the safety/tolerability of increased erlotinib doses; to evaluate the incidence of grade ⩾2 rash with erlotinib dose escalation; to compare progression-free survival (PFS), ORR, and disease control rates (DCRs) between randomised arms; to make a non-randomised comparison of efficacy/safety between patients who developed grade 0 or 1 vs grade ⩾2 rash during the 4-week run-in period. The per-protocol trigger for analysis was 120 deaths.
Management of rash
Strategies for rash management in the RACHEL study are described in the Supplementary Materials (Supplementary Table S1).
Biomarker analyses
Exploratory objectives included the correlation of EGFR protein expression, EGFR gene copy number, KRAS and EGFR mutations, and EGFR intron 1 polymorphisms with efficacy (Supplementary Appendix 1).
Statistical analyses
The intent-to-treat (ITT) population included all patients in the randomised treatment arms (standard dose/dose escalation), excluding patients ineligible for randomisation following the run-in period. To detect a survival HR of 0.6 between randomised arms (80% power, two-sided 5% significance) 120 events were required. Assuming 24 months’ accrual, 9 months’ follow-up, and a 5% drop out rate per year, 70 patients were required per randomised arm (requiring 560 patients to be enrolled). The hypothesis was that OS and PFS would be statistically significantly different between the standard-dose and dose-escalation arms. OS and PFS from randomisation were analysed in the ITT population using a two-sided log-rank test (α=0.05) and stratified log-rank tests; OS and PFS from the start of treatment were analysed using non-stratified log-rank tests. Median and 95% CI from randomisation were estimated using the Kaplan–Meier methodology. The OS and PFS of patients in the non-randomised grade ⩾2 rash arm were measured from the start of treatment. The OS and PFS in the non-randomised grade ⩾2 rash and the randomised arms were compared using a two-sided log-rank test (α=0.05; no adjustment for multiple comparisons). The Cox proportional hazards model was used to estimate the HR and 95% CI of OS/PFS (from the start of treatment). ORR and DCR (and 95% CI) were evaluated using the Pearson–Clopper methodology. The rates in the randomised arms were compared by χ2-tests (α=0.05; no adjustment for multiple comparisons). The 95% CI for the rate difference between the two arms was calculated using the Hauck–Anderson approach. The safety population included all patients who received at least one dose of trial medication and had at least one safety follow-up.
Results
Patient population
A total of 467 patients were enrolled (May 2008 to May 2010). Of these, 36 patients were classed as early drop outs and 285 were not eligible for randomisation as they had grade ⩾2 rash (n=106) or they had grade 0 or 1 rash but did not meet other inclusion criteria for randomisation (n=179). The full disposition of patients through the study to the data cutoff for the primary analysis (19 May 2011) is detailed (Figure 2). Baseline characteristics (Table 1) were generally well matched between the randomised arms. More patients in the dose-escalation arm, compared with the standard-dose arm, had an ECOG PS 1, pancreatic cancer pain, and tumour location in the pancreas tail. The non-randomised grade ⩾2 arm was reasonably well balanced with the randomised arms, with the exception that there were fewer current smokers in the non-randomised arm.

CONSORT diagram of patient disposition through the study. Standard dose: patients with grade 0 or 1 rash after the run-in period and eligible for randomisation (no PD and no dose adjustments to <75% of the full dose of chemotherapy). Dose escalation: patients with grade 0 or 1 rash after the run-in period and eligible for randomisation (no PD and no dose adjustments to <75% of the full dose of chemotherapy). Non-randomised, non-eligible: patients with grade 0 or 1 rash after the run-in period, but non-eligible for randomisation (having PD or dose adjustments to <75% of the full dose of chemotherapy). Non-randomised, grade ⩾2 rash: patients with grade ⩾2 rash after the run-in period. Early dropouts: patients with ECOG PS 2, PD, or serious adverse event leading to treatment discontinuation or death before randomisation. Abbreviations: ECOG PS=Eastern Cooperative Oncology Group performance status; ITT=intent-to-treat; PD=disease progression. aOne patient excluded due to dose escalation before randomisation. bTwo patients randomised to the dose-escalation arm, but who did not receive dose escalation were added. cTwo patients randomised to the dose-escalation arm, but who did not receive dose escalation were removed and two patients with dose escalation from the non-randomised arm were added.
The median follow-up was 17.4 months (range, 1.1–24.0) in the standard-dose arm and 20.9 months (range, 0.0–30.4) in the dose-escalation arm. At the cut-off, 61 (81.3%) patients in the standard-dose arm, 60 (85.7%) patients in the dose-escalation arm, and 90 (84.9%) patients in the non-randomised grade ⩾2 rash arm had died.
A total of 7 out of 75 (9.3%) patients developed a grade ⩾2 rash in the standard-dose arm, compared with 29 out of 70 (41%) patients in the dose-escalation arm (Supplementary Table S2). The median time from randomisation to onset of grade ⩾2 rash was 71 days (range, 6–347) in the standard-dose arm and 28 days (range, 2–537) in the dose-escalation arm (Supplementary Table S2).
Efficacy outcomes
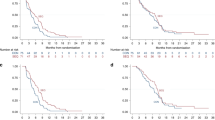
OS from randomisation was not statistically different between the standard-dose and dose-escalation arms (HR, 1.26, 95% CI: 0.88–1.80; P=0.2026; median OS: 8.4 vs 7.0 months, respectively; Figure 3). Similar results were observed when OS was analysed by the stratification factor (Supplementary Figure S1). PFS from randomisation was not significantly different between the standard-dose and dose-escalation arms (HR, 1.09, 95% CI: 0.77–1.54; P=0.6298; median PFS: 4.5 vs 3.5 months, respectively). The ORR and DCR were similar between the standard-dose and dose-escalation arms (ORR: 14.7% vs 8.6%, respectively; DCR: 62.7% vs 47.1%, respectively; P=0.0603; Table 2). No prespecified biomarker was found to be predictive of a better outcome (Supplementary Appendix 1; Supplementary Figure S2). OS from the start of treatment was not statistically different between the non-randomised grade ⩾2 arm and the standard-dose arm (HR, 0.83; 95% CI: 0.60–1.15; P=0.2678; median OS: 7.9 vs 9.3 months, respectively) or the dose-escalation arm (HR, 1.03; 95% CI: 0.74–1.43; P=0.8449; median OS: 7.9 vs 8.0 months, respectively), though the study was insufficiently powered to detect differences (Figure 4). PFS from the start of treatment differed significantly in the non-randomised grade ⩾2 arm vs the standard-dose arm (HR, 0.69; 95% CI: 0.51–0.95; P=0.0217; median PFS: 4.0 vs 5.4 months, respectively) but not vs the dose-escalation arm (HR, 0.79; 95% CI: 0.57–1.10; P=0.1596; median PFS: 4.0 vs 4.5 months, respectively).

OS (from randomisation) for patients who were randomised to the standard therapy arm (gemcitabine plus erlotinib 100 mg per day) or the dose-escalation arm (gemcitabine plus escalating doses of erlotinib). Abbreviations: HR=hazard ratio; OS=overall survival.

OS (from the start of the run-in period) for patients who did not develop grade ⩾2 rash during the 4-week run-in period who were randomised to receive standard therapy (gemcitabine plus erlotinib 100 mg per day) or dose escalation (gemcitabine plus escalating doses of erlotinib), compared with non-randomised patients who developed grade ⩾2 rash during the run-in period. Abbreviation: OS=overall survival.
Pharmacokinetics
The population pharmacokinetics were within the range of that described by the existing pharmacokinetic model and are discussed in Supplementary Appendix 2.
Safety and tolerability
Most patients received between 1 and 8 months of erlotinib (range, 0–17 months) and between 2 and 11 gemcitabine cycles (range, 2–19). Cumulative doses are reported in the Supplementary Materials (Supplementary Table S3). In the dose-escalation arm, 1 (1%) patient remained on 100 mg per day erlotinib, 70 (99%) patients received 150 mg per day, 49 (69%) received 200 mg per day, 28 (39%) received 250 mg per day and 1 (1%) patient received 300 mg per day. The main reason for treatment withdrawal in any arm was insufficient therapeutic response (Supplementary Table S4). Dose reductions/interruptions are also reported (Supplementary Table S5).
All patients experienced ⩾1 AE, with the exception of three patients in the standard-dose arm (Table 3). The most frequent AEs were skin/subcutaneous tissue disorders. Of the non-randomised patients who experienced grade ⩾2 rash during the run-in period, 8.6% had a serious AE (SAE) leading to death compared with 2.6% of patients in the randomised standard-dose arm and 2.8% of patients in the dose-escalation arm. In addition, 73.3% of non-randomised patients who experienced grade ⩾2 rash had severe AEs, compared with 53.2% of patients in the standard-dose arm and 66.2% of patients in the dose-escalation arm. In the randomised population, the incidence of SAEs was comparable between treatment arms (standard dose 31% vs dose escalation 28%). Treatment-related AEs were more common in the dose-escalation and the non-randomised grade ⩾2 rash arms (both 100%) compared with the standard-dose arm (87%). Treatment-related SAEs were comparable in the standard-dose (13%) and dose-escalation arms (10%). The most common SAEs were gastrointestinal disorders and infections/infestations, and the most common grade 3 or 4 AE was neutropenia (Table 4); frequencies were generally comparable between treatment arms with the exception of gastrointestinal disorders and neutropenia, which were seen slightly more often in non-randomised patients (grade ⩾2 rash).
Discussion
Of patients in the erlotinib dose-escalation arm, 41% developed grade ⩾2 rash, in contrast to 9.3% of patients in the standard-dose arm. Why only some patients develop rash may be related to drug exposure differences, which may, in turn, be related to genetic mechanisms. EGFR polymorphism (specifically a small number of EGFR intron 1CA dinucleotide repeats) is associated with increased EGFR expression and rash (Amador et al, 2004). The ability of dose escalation to induce rash is suggestive of a dose-response relationship; however, this relationship did not extend to efficacy. There was no difference in OS or PFS in the dose-escalation arm compared with the standard-dose arm. The efficacy results are not in line with the expected outcome and the results of previous pancreatic cancer studies. Improved survival was reported for erlotinib-treated patients who developed a grade ⩾2 vs 0 rash in the phase III PA.3 study (OS: median 10.8 vs 5.4 months, respectively; HR, 0.47, 95% CI: 0.34–0.64; P<0.001; PFS: median 6.5 vs 3.1 months, respectively; HR, 0.46, 95% CI: 0.33–0.65; P<0.001) (Wacker et al, 2007). In addition, a phase II study of erlotinib plus gemcitabine reported better outcomes for patients with grade ⩾2 rash compared with grade <2 rash (OS: median 11 vs 5 months, respectively; P<0.001; PFS: median 6 vs 3 months, respectively; P<0.001; ORR: 21% vs 7%, respectively; P<0.05; DCR: 84% vs 43%, respectively; P<0.05) (Aranda et al, 2012). The AViTA study reported a numerically longer OS for gemcitabine and erlotinib-treated patients who developed a grade ⩾2 rash compared with those with no rash (median 8.4 vs 5.0 months) (Verslype et al, 2009).
Retrospective analyses have also highlighted relationships between rash and efficacy in pancreatic cancer. Patients treated with erlotinib plus gemcitabine classified as having ‘high’ severity rash had significantly longer OS/PFS than their ‘low’ severity counterparts (OS: median 7.58 vs 5.03 months; P=0.0339; PFS: median 2.4 vs 2.0 months; P=0.0485) (Stepanski et al, 2013). Another retrospective study indicated that erlotinib-induced grade 1 and grade ⩾2 rash were correlated with improved OS (HR, 0.71, 95% CI: 0.50–1.00 and HR, 0.57, 95% CI: 0.40–0.82, respectively; P=0.007), although adjusting for baseline characteristics removed the significance (HR, 0.69, 95% CI: 0.49–0.97 and adjusted HR, 0.78, 95% CI: 0.48–1.2, respectively) (Alejandro et al, 2012). In the present study, the slight tendency of dose-escalated patients to have negative prognostic factors (ECOG PS 1; pancreatic cancer pain; pancreatic tail tumours) may have contributed to the lack of difference seen between the randomised groups.
The key difference between the RACHEL trial and previous pancreatic cancer trials was the aspect of using erlotinib dose escalation to induce rash. A phase II NSCLC study, a disease where a relationship with rash is well established, employed the ‘dose-to-rash’ strategy and also demonstrated no correlation of erlotinib exposure with tumour response (Mita et al, 2011). In RACHEL, the OS was also not statistically different between the group of patients who developed a grade ⩾2 rash ‘naturally’ after 4 weeks, and those in whom rash was induced by dose escalation (HR, 1.03, 95% CI: 0.74–1.43; P=0.8449). However, the patients with ‘natural’ grade ⩾2 rash were not randomised, and the analysis was not adjusted for baseline characteristics. Although the rash may appear be more severe in the non-randomised grade ⩾2 ‘natural’ rash group (18.1% grade 3 or 4 rash, compared with 4.2% and 0% in the two randomised groups), this is not surprising, given that the non-randomised group will have a greater probability of grade 3 or 4 rash by virtue of their randomisation criteria (all grade ⩾2 rash).
Increased awareness of rash by patients and physicians means that symptoms are treated earlier and more effectively than in earlier studies. A rash, which (due to poor awareness or sub-optimal management) may have developed to grade ⩾2 in older studies, may have been treated earlier and more effectively in RACHEL. This may have contributed to correlations between rash and efficacy not being observed in this study, in contrast to older studies.
The results should also be considered in light of cumulative exposure to erlotinib and gemcitabine. The mean erlotinib exposure was higher in the dose-escalation arm, compared with the randomised standard-dose arm, as expected. The mean erlotinib dose was lowest in the non-randomised grade ⩾2 rash arm, due to a higher rate of erlotinib dose reduction/interruptions (relative to the standard-dose arm). The mean exposure to gemcitabine was lower in the dose-escalation arm than the standard-dose arm, despite similar proportions of patients having dose interruptions/modifications/withdrawals. PFS was shorter in the dose-escalation arm, and therefore, gemcitabine exposure may be lower than expected due to PD (rather than dose-limiting toxicity). Similar numbers of patients withdrew from trial treatment between the randomised arms owing to safety reasons (AEs/death) or insufficient therapeutic response.
Although RACHEL was unable to prospectively demonstrate a survival benefit with erlotinib for patients with an induced grade ⩾2 rash, taken in the context of previous studies, rash and efficacy of EGFR TKIs do appear to be linked. EGFR TKI rash generation stems from high EGFR levels in the skin basal layer/hair follicle root sheaths. EGFR inhibition slows epithelial cell regeneration and reduces chemokine expression control. Altered epidermal/stratum corneum growth and differentiation leads to damage of the sebaceous glands and follicular epithelium. Inflammatory mediators are released producing an abnormal skin structure primed for bacterial colonisation, triggering further inflammatory reactions (Guttman-Yassky et al, 2010). This can result in acneiform rash (papules/pustules), xeroderma (dry skin/scaling/fissuring), pruritus (and lichenification), paronychia, tenderness, pain, and secondary infection. Strategies to manage skincare are summarised elsewhere (Saif et al, 2008).
As expected, skin toxicity was the most common AE in RACHEL. No new safety signals were observed. The dose-escalation arm reported more treatment-related AEs, and the standard-dose arm had more treatment-related SAEs, but the incidence of patients withdrawn due to toxicity was comparable between the arms. Differences in gemcitabine or erlotinib exposure may have contributed to differences in AEs/SAEs.
In conclusion, dose escalation of erlotinib was able to induce rash in a subset of patients who did not develop grade ⩾2 rash during a 4-week run-in period of standard erlotinib plus gemcitabine. There was no evidence that the higher dose translated into increased benefit for patients. Therefore, there is no recommendation to increase the dose of erlotinib beyond that approved for patients with metastatic pancreatic cancer.
Change history
25 November 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alejandro LM, Adel NG, O’Reilly EM, Riedel E, Lacouture ME (2012) Association between skin toxicities and treatment outcomes in patients with pancreatic cancer (PC) receiving erlotinib (E): Memorial Sloan-Kettering Cancer Center (MSKCC) experience. J Clin Oncol 30: Abstract 294.
Amador MR, Oppenheimer D, Perea S, Maitra A, Cusatis G, Iacobuzio-Donahue C, Baker SD, Ashfaq R, Takimoto C, Forastiere A, Hidalgo M (2004) An epidermal growth factor receptor intro 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res 64: 9139–9143.
Aranda E, Manzano JL, Rivera F, Galán M, Valladares-Ayerbes M, Pericay C, Safont MJ, Mendez MJ, Irigoyen A, Arrivi A, Sastre J, Díaz-Rubio E (2012) Phase II open-label study of erlotinib in combination with gemcitabine in unresectable and/or metastatic adenocarcinoma of the pancreas: relationship between skin rash and survival (Pantar study). Ann Oncol 23: 1919–1925.
Buges C, Marti AM, Rosell R, Vergnenegre A, De Marinis F, Massuti B, De Castro J, Gervais R, Costa EC, Moran T, Santarpia M, Felip E, Majem M, Porta R, Palmero R, Drozdowskyj A, Heidecke C, Gasco A, Taron M, Paz-Ares LG (2012) Skin toxicity associated with outcome to erlotinib in non-small cell lung cancer (NSCLC) patients (p) with EGFR mutations in the EURTAC study. J Clin Oncol 30: Abstract 7542.
Burris H 3rd, Rocha-Lima C (2008) New therapeutic directions for advanced pancreatic cancer: targeting the epidermal growth factor and vascular endothelial growth factor pathways. Oncologist 13: 289–298.
Clark GM, Perez-Soler R, Sui L, Gordon A, Santabárbara P (2003) Rash severity is predictive of increased survival with erlotinib HCl. Proc Am Soc Clin Oncol 22: Abstract 786.
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825.
da Cunha Santos G, Dhani N, Tu D, Chin K, Ludkovski O, Kamel-Reid S, Squire J, Parulekar W, Moore MJ, Tsao MS (2010) Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA.3. Cancer 116: 5599–5607.
Guttman-Yassky E, Mita A, De Jonge M, Matthews L, McCarthy S, Iwata KK, Verweij J, Rowinsky EK, Krueger JG (2010) Characterisation of the cutaneous pathology in non-small cell lung cancer (NSCLC) patients treated with the EGFR tyrosine kinase inhibitor erlotinib. Eur J Cancer 46: 2010–2019.
Merl MY, Abdelghany O, Li J, Saif MW (2010) First-line treatment of metastatic pancreatic adenocarcinoma: can we do better? JOP 11: 317–320.
Mita AC, Papadopoulos K, de Jonge MJ, Schwartz G, Verweij J, Mita MM, Ricart A, Chu QS, Tolcher AW, Wood L, McCarthy S, Hamilton M, Iwata K, Wacker B, Witt K, Rowinsky EK (2011) Erlotinib ‘dosing-to-rash': a phase II intrapatient dose escalation and pharmacologic study of erlotinib in previously treated advanced non-small cell lung cancer. Br J Cancer 105: 938–944.
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W National Cancer Institute of Canada Clinical Trials Group (2007) Erlotinib plus gemcitabine compared with gemcitabine along in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Trials Group. J Clin Oncol 25: 1960–1966.
Peeters M, Siena S, Van Custem E, Sobrero A, Hendlisz A, Cascinu S, Kalofonos H, Devercelli G, Wolf M, Amado RG (2009) Association of progression-free survival, overall survival, and patient-reported outcomes by skin toxicity and KRAS status in patients receiving panitumumab monotherapy. Cancer 115: 1544–1554.
Perez-Soler R, Leon L, Wojtowicz-Praga S (2012) Cutaneous toxicity secondary to erlotinib therapy in patients with non-small cell lung cancer in the NCIC CTG BR.21 study: time course and correlation with survival. J Clin Oncol 30: Abstract 7573.
Philip PA, Benedetti J, Fenoglio-Preiser C, Zalupski M, Lenz H, O'Reilly E, Wong R, Atkins J, Abruzzese J, Blanke C (2007) Phase III study of gemcitabine [G] plus cetuximab [C] versus gemcitabine in patients [pts] with locally advanced or metastatic pancreatic adenocarcinoma [PC]: SWOG S0205 study. J Clin Oncol 25: Abstract LBA4509.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, de Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Muñoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, de Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13: 239–246.
Saif MW, Kerikas I, Tsmiboukis S, Syrigos K (2008) Erlotinib-induced skin rash. Pathogenesis, clinical significance and management in pancreatic cancer patients. JOP 9: 267–274.
Saltz L, Kies M, Abbruzzese L, Needle M (2003) The presence and intensity of the cetuximab-induced acne-like rash predicts increased survival in studies across multiple malignancies. Proc Am Soc Clin Oncol 22: Abstract 817.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabárbara P, Seymour L National Cancer Institute of Canada Clinical Trials Group (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353: 123–132.
Stepanski EJ, Reyes C, Walker MS, Satram-Hoang S, Leon L, Wojtowicz-Praga S, Miller PJ, Houts AC, Schwartzberg LS (2013) The association of rash severity with overall survival: findings from patients receiving erlotinib for pancreatic cancer in the community setting. Pancreas 42: 32–36.
Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y, Oida Y, Yamazaki H, Nakamura M, Ueyama Y, Tanaka M, Inokuchi S, Makuuchi H (2003) Epidermal growth factor receptor expression in human pancreatic cancer: significance for liver metastasis. Int J Mol Med 11: 305–309.
Van Cutsem E, Vervenne WL, Bennouna J, Humblet Y, Gill S, Van Laethem JL, Verslype C, Scheithauer W, Shang A, Cosaert J, Moore MJ (2009a) Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol 27: 2231–2237.
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009b) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. New Eng J Med 360: 1408–1417.
Van Cutsem E, Tejpar S, Vanbeckevoort D, Peeters M, Humblet Y, Gelderblom H, Vermorken JB, Viret F, Glimelius B, Gallerani E, Hendlisz A, Cats A, Moehler M, Sagaert X, Vlassak S, Schlichting M, Ciardiello F (2012) Intrapatient cetuximab dose escalation in metastatic colorectal cancer according to the grade of early skin reactions: the randomized EVEREST study. J Clin Oncol 30: 2861–2868.
Verslype C, Vervenne W, Bennouna J, Humblet Y, Cosaert J, Van Cutsem E (2009) Rash as a marker for efficacy of gemcitabine plus erlotinib-based therapy in pancreatic cancer: results from the AViTA study. J Clin Oncol 27: Abstract 4532.
Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ (2007) Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res 13: 3913–3921.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L, You C (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12: 735–742.
Acknowledgements
We would like to thank all patients who participated in the study and clinical personnel involved in data collection. The trial was funded by the study sponsor, F Hoffmann-La Roche, Basel, Switzerland, who also provided support for third-party writing assistance for this manuscript, from Emma McConnell of Gardiner-Caldwell Communications.
Author contributions
CH, BK, and C-PL designed the study. EVC developed protocols. IF, EN, and J-LVL recruited patients. GA, TC, IF, CH, BK, C-PL, SMS, RL, MM, EN, and CKT collected data. ZC, CH, SMS, BK, C-PL, XM, MM, AZ, and EVC analysed the data. GA, TC, ZC, AF, CH, BK, C-PL, RL, XM, EN, J-LVL, AZ, SMS, and EVC interpreted the data. TC and C-PL performed the literature search. All authors contributed to the drafting or revising of the manuscript, and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
AF, BK, and AZ are all employees of F. Hoffmann-La Roche Ltd. RL has received honoraria from F. Hoffmann-La Roche Ltd. for lectures and projects. MM has received funding for clinical trials from F. Hoffmann-La Roche Ltd. EVC has received research funding for his institution from F. Hoffmann-La Roche. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Van Cutsem, E., Li, CP., Nowara, E. et al. Dose escalation to rash for erlotinib plus gemcitabine for metastatic pancreatic cancer: the phase II RACHEL study. Br J Cancer 111, 2067–2075 (2014). https://doi.org/10.1038/bjc.2014.494
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.494
Keywords
This article is cited by
-
Predictive Value of Early Skin Rash in Cetuximab-Based Therapy of Advanced Biliary Tract Cancer
Pathology & Oncology Research (2018)
-
A phase II trial of erlotinib monotherapy in advanced pancreatic cancer as a first- or second-line agent
Cancer Chemotherapy and Pharmacology (2017)
-
Pancreatic cancer: from state-of-the-art treatments to promising novel therapies
Nature Reviews Clinical Oncology (2015)

{kind=link}
{kind=link}
{kind=link}