
Abstract
Background:
The role of tumour-infiltrating inflammation in the prognosis of patients with colorectal cancer (CRC) has not been fully evaluated. The primary objective of our meta-analysis was to determine the impact of tumour-infiltrating inflammation on survival outcomes.
Methods:
Ovid MEDLINE and EMBASE were searched to identify studies reporting the prognostic significance of tumour-infiltrating inflammation for patients with CRC. The primary outcome measures were overall survival (OS), cancer-specific survival (CS) and disease-free survival (DFS).
Results:
A total of 30 studies involving 2988 patients were identified. Studies were subdivided into those considering the associations between CRC survival and generalised tumour inflammatory infiltrate (n=12) and T lymphocyte subsets (n=18). Pooled analyses revealed that high generalised tumour inflammatory infiltrate was associated with good OS (HR, 0.59; 95% CI, 0.48–0.72), CS (HR, 0.40; 95% CI, 0.27–0.61) and DFS (HR, 0.72; 95% CI, 0.57–0.91). Stratification by location and T lymphocyte subset indicated that in the tumour centre, CD3+, CD8+ and FoxP3+ infiltrates were not statistically significant prognostic markers for OS or CS. In the tumour stroma, high CD8+, but not CD3+ or FoxP3+ cell infiltrates indicated increased OS. Furthermore, high CD3+ cell infiltrate was detected at the invasive tumour margin in patients with good OS and DFS; and high CCR7+ infiltrate was also indicated increased OS.
Conclusion:
Overall, high generalised tumour inflammatory infiltrate could be a good prognostic marker for CRC. However, significant heterogeneity and an insufficient number of studies underscore the need for further prospective studies on subsets of T lymphocytes to increase the robustness of the analyses.
Similar content being viewed by others


Main
The clinical guidelines for the treatment of colorectal cancer (CRC) are mainly based on the TNM classification scheme proposed by the American Joint Committee on Cancer. However, the current staging system has not been adequately validated for treatment planning and prognosis assessments. According to the guidelines, adjuvant therapy is not recommended for patients with stage I or low-risk stage II CRC after radical surgery because, theoretically, those patients can be completely cured and can achieve long-term survival (Benson et al, 2013). However, approximately 10% of stage I and 20% of stage II patients experience recurrence or metastasis. Moreover, pronounced heterogeneity in survival outcome is noted among patients with stage II CRC, which accounts for approximately 40% of all CRC cases. For those patients, TNM stage cannot serve as an early warning marker for postoperative metastasis or recurrence. Therefore, additional accurate prognostic and predictive markers for staging must be identified as a complement to the TNM system for postoperative adjuvant therapy.
The local tumour microenvironment, comprises tumour cells, extracellular matrix, immune cells, cytokines and other factors, has an important role in tumour formation, growth, invasion and metastasis. Immune cells, particularly T lymphocytes, serve as regulatory factors in the tumour microenvironment (Liotta and Kohn, 2001; Li et al, 2007). Jass (1986) first proposed that infiltration of immune cells is a novel independent prognostic factor in CRC, and this new system was considered superior to the Dukes’ staging system. Using molecular biological methods, such as immunohistochemistry (IHC) and haematoxylin-eosin (HE) staining, various studies have demonstrated that the type of immune cells (e.g., CD3+/CD8+/FOXP3+ T lymphocytes) and the density or location of tumour-infiltrating T cells have a prognostic correlation for CRC (Naito et al, 1998; Chiba et al, 2004; Galon et al, 2006; Salama et al, 2009; Sinicrope et al, 2009; Correale et al, 2010; Deschoolmeester et al, 2010; Chew et al, 2011). Such a correlation has been well described in a variety of tumours, such as ovarian cancer and breast cancer (Sato et al, 2005; Tomsova et al, 2008; Mahmoud et al, 2011; Liu et al, 2012). However, the prognostic value of tumour-infiltrating lymphocytes in CRC remains controversial because of the limited number of studies.
To date, results have varied greatly among studies because of differences in their design, assay methods and reported outcomes, making it difficult to interpret the overall estimates. Hence, based on the published studies, we performed a systematic review and meta-analysis to investigate the relationship between tumour inflammatory infiltrates and CRC survival stratified according to T lymphocyte subset and infiltration site.
Materials and methods
Search strategy
The Ovid MEDLINE (1946 to August 2013) and EMBASE (1976 to August 2013) databases were searched without limits to identify all relevant studies. The literature search included Medical Subject Headings and Emtree headings and related text and keyword searches in a manner that combined terms related to the prognostic effect of tumour inflammatory infiltration on patients with CRC. Detailed search terms and strategies for both databases are provided in Supplementary Appendix 1. We also explored reference lists of previously published reviews and meta-analyses. We did not include conference abstracts in the analysis because of the insufficient data provided by the authors.
Study selection and inclusion criteria
Two independent reviewers (ZBM and YL or AC) selected the identified studies based on the title and abstract. If the study’s topic could not be ascertained from its title or abstract, the full-text version would be retrieved for evaluation. Disagreement was resolved by discussion or consensus or with a third party (CYL). Original studies were eligible for inclusion if all patients were surgically treated for primary CRC. Studies were included if they focused on generalised tumour inflammatory infiltrate and associated T lymphocyte subsets (including CD3+, CD8+, FoxP3+, CCR7+, CD45RO+ and GRB+ lymphocytes) in CRC patients identified by HE staining and/or IHC staining (high versus low density) and reported prognostic information, including overall survival (OS), cancer-specific survival (CS) and/or disease-free survival (DFS). We included the analysis of lymphocytes in the tumour centre (CC) and tumour stroma (ST) and at the invasive tumour margin (IM). Exclusion criteria included the following: patients had autoimmune diseases, <30 patients were considered in the analysis or insufficient data were provided to calculate the hazard ratio (HR) with a 95% confidence interval (CI). Review articles, case reports, commentaries and letters were not included. However, references from those forms of articles were manually searched to identify additional relevant studies. In studies with multiple publications, only data from the most recent publication were included in the meta-analysis. If possible, authors of included studies were contacted for unreported data.
Data extraction
Two reviewers (ZBM and YL or AC) independently selected articles and extracted data from eligible studies. A standardised data abstraction form was developed, and key elements pertaining to the study design, sample size, patient age, stage of disease, T lymphocyte subset, T lymphocyte counting site, follow-up duration, use of multivariate logistic model analysis, adjustment variables, HR estimates (with the corresponding 95% CIs) for the high density over the low density of each T-cell subset at certain locations within tumours (CT, ST or IM) and the HR cutoff point were obtained. Discrepancies were resolved by discussion by a third reviewer (CYL) until the two reviewers reached consensus or by contacting content experts if necessary.
Study quality and risk of bias assessment
The quality of each study was assessed using an established form that was first developed and applied by McShane et al (2005) and Hayes et al (1996) (Supplementary Appendix 3). The following seven domains were assessed and scored on a scale from 0 to 8: inclusion and exclusion criteria, study design (prospective or retrospective), patient and tumour characteristics, description of the method or assay, study endpoints, follow-up time with patients and number of patients that dropped out during the follow-up period.
Subgroup analyses and definition of prognostic outcomes
We conducted subgroup analyses to investigate associations between prognostic outcomes (OS, CS and DFS) and both T lymphocyte subsets (CD3+, CD8+, FoxP3+ and CCR7+ lymphocytes) and T lymphocyte infiltrate location (CT, ST or IM). Overall, survival was defined as the time elapsing from the date of initial primary diagnosis of CRC to the date of death irrespective of the cause of death. CS was defined as the interval between the initial primary diagnosis of CRC and the last objective follow-up information or death caused by the disease. DFS was defined as the interval between the date of initial primary diagnosis of CRC and the date of the first relapse or death.
Statistical analysis
We used Stata statistical software (version 12.0; Stata Corporation, College Station, TX, USA) to perform the meta-analysis. For time-to-event outcomes, we retrieved the HR estimates with a 95% CI from the original articles. When Kaplan–Meier curves were provided rather than the HR, the HR was estimated indirectly from the curves using the methods described by Parmar et al (1998 and Tierney et al (2007).
The HR abstracted in each study provided an estimate of the ratio of the HR for high-density over low-density tumour inflammatory infiltrate. Thus, an HR <1 favoured high density and implied a good prognosis, whereas an HR >1 favoured low density and implied a poor prognosis. We then performed subgroup analyses according to the type and location of the T lymphocyte infiltrates. Summary statistics were derived with a random-effect model because some evidence for heterogeneity across the studies was observed in the meta-analysis (e.g., study designs, methods and duration of follow-up), and HRs and 95% CIs were pooled regarding related outcomes according to the DerSimonian and Laird methods (Borenstein et al, 2011). Interstudy heterogeneity was quantified using the I2 statistic, with an I2 value >50% indicating substantial heterogeneity. Potential sources of heterogeneity were then investigated using a predefined form (Supplementary Appendix 3) in some domains reported by de Graeff et al (2009).
In addition, subgroup analyses were performed on each T lymphocyte subset according to infiltration location. Also, the most frequently reported subsets, including generalised tumour inflammatory infiltrate and CD3+, CD8+, FoxP3+ and CCR7+ lymphocytes, were assessed.
Publication bias was examined visually by inspecting funnel plots and statistically by using Egger’s or Begg’s regression model (Egger et al, 1997), in which a P-value of <0.10 was considered significant (Hedges et al, 1985; Higgins and Thompson, 2002). Reasons for statistical heterogeneity were explored through sensitivity analyses.
Results
Literature search
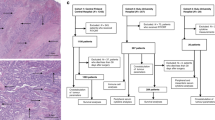
Figure 1 presents the selection process of the eligible studies. In brief, among the 2988 studies identified for initial evaluation, 32 studies were eligible for further assessment. Two studies had published insufficient data (Liska et al, 2011; Zlobec et al, 2008) (i.e., did not provide a HR with a 95% CI or data sufficient for estimating the HR). We tried to contact the corresponding authors of these two studies for additional information, but neither of them responded. Therefore, 30 studies were left with sufficient data for extraction.

Flowchart of the study selection.
Study characteristics
The baseline characteristics of each study are summarised in Supplementary Appendix 2 (Ropponen et al, 1997; Naito et al, 1998; Nielsen et al, 1999; Guidoboni et al, 2001; Nagtegaal et al, 2001; Cianchi et al, 2002; Chiba et al, 2004; Menon et al, 2004; Buckowitz et al, 2005; Gao et al, 2005; Günther et al, 2005; Klintrup et al, 2005; Schimanski et al, 2005; Galon et al, 2006; Szynglarewicz et al, 2007; Ogino et al, 2009; Roxburgh et al, 2009; Salama et al, 2009; Sinicrope et al, 2009; Correale et al, 2010; Deschoolmeester et al, 2010; Frey et al, 2010; Lee et al, 2010; Nosho et al, 2010; Simpson et al, 2010; Correale et al, 2012; Huh et al, 2012; Richards et al, 2012; Yoon et al, 2012; Kim et al, 2013). Generalised tumour inflammatory infiltrates were reported in 12 studies (Cianchi et al, 2002; Ropponen et al, 1997; Nielsen et al, 1999; Nagtegaal et al, 2001; Buckowitz et al, 2005; Gao et al, 2005; Klintrup et al, 2005; Szynglarewicz et al, 2007; Ogino et al, 2009; Roxburgh et al, 2009; Huh et al, 2012; Richards et al, 2012) and 15 studies included T lymphocyte subsets (Naito et al, 1998; Guidoboni et al, 2001; Chiba et al, 2004; Menon et al, 2004; Günther et al, 2005; Schimanski et al, 2005; Galon et al, 2006; Salama et al, 2009; Sinicrope et al, 2009; Correale et al, 2010; Deschoolmeester et al, 2010; Frey et al, 2010; Lee et al, 2010; Nosho et al, 2010; Simpson et al, 2010; Correale et al, 2012; Yoon et al, 2012; Kim et al, 2013). The majority of the studies were performed in Europe (n=21). Others were conducted in Asia (n=4), North America (n=4) and Oceania (n=1). The total sample size from all studies was 7840, with a mean of 261 patients (ranging from 45 to 843 patients) per study. Ten studies included fewer than 100 patients and 15 studies enrolled >200 patients. All studies were published between 1997 and 2013.
The eligible studies applied either IHC (n=18) or HE staining (n=12) as a detection method. The study design variables and summary results are provided in Tables 1 and 2, respectively. The assessment of risk of bias for the individual studies indicated that 16 studies had quality scores of >4, whereas the remaining 14 studies had scores of 4 (n=9) or <4 (n=5). Analyses of tumour inflammatory lymphocyte type and lymphocyte subset as prognostic factors were confirmed in a multivariate analysis in 24 of the included studies. HRs for OS, CS or DFS could be directly extracted in 25 studies and were estimated through survival curves for the four remaining studies (Chiba et al, 2004; Günther et al, 2005; Schimanski et al, 2005; Galon et al, 2006; Lee et al, 2010; Kim et al, 2013).
The most frequently used cutoff values for the high versus low/present versus absent density of tumour-infiltrating lymphocytes were the median (n=8) and values calculated using several semiquantitative methods, including K-M criteria (scores of 0 and 1 versus scores of 2 and 3; n=4) and Jass classification (none/few versus extensive; n=4) (see Supplementary Appendix 2 for details).
Subgroup analysis
Subgroup analyses were performed and the results are summarised in Table 2. We first evaluated the prognostic significance of generalised tumour inflammatory infiltrates evaluated by general counts of tumour-infiltrating lymphocytes in CRC and some of the most frequently studied T lymphocyte subsets stratified by tumour location (CT, ST or IM). Estimation of between-study heterogeneity was highly imprecise because of the limited number and low individual power of studies available in each subgroup. Therefore, funnel plot analyses were performed only for subsets of generalised tumour inflammatory infiltrates and T lymphocyte subgroups in the tumour centre for OS that included a relatively high number of studies. Furthermore, we analysed the influence of study quality, tumour stage, study design, sample size, cutoff criteria and duration of follow-up on the pooled results for OS for the above subset.
Generalised tumour inflammatory infiltrate
Twelve studies were pooled for analysis of the density of generalised tumour inflammatory infiltrates at the invasive margin. All studies indicated a good prognostic effect for OS (HR=0.59; 95% CI, 0.48–0.72), CS (HR=0.40; 95% CI, 0.27–0.61) and DFS (HR=0.72; 95% CI, 0.57–0.91; Figure 2A). Egger’s test (P=0.963) and Begg’s test (P=1.00), as well as funnel plots, provided no evidence of publication bias (Figure 3A) for OS. However, moderate heterogeneity was noted (I2=26.8%, P=0.206). Therefore, we performed subgroup analyses to evaluate the effect of the interstudy variability on the pooled results (see Table 3), and these analyses revealed that high levels of generalised tumour inflammatory infiltrate were associated with improved OS in studies with high study quality (score >4; HR=0.55; 95% CI, 0.43–0.71) and large sample size (⩾200; HR=0.56; 95% CI, 0.44–0.71); however, statistical significance was not noted in studies with low study quality (HR=0.78; 95% CI, 0.49–1.23) or small sample size (HR=0.84; 95% CI, 0.46–1.56). Similarly, subgroup analyses indicated that for this subset, prognostic benefit was observed for both stages I–III CRC (HR=0.43; 95% CI, 0.26–0.72) and stages I–IV CRC (HR=0.66; 95% CI, 0.55–0.79); prospective studies (HR=0.54; 95% CI, 0.37–0.78) and retrospective studies (HR=0.64; 95% CI, 0.51–0.81); standard cutoff criteria (HR=0.57; 95% CI, 0.39–0.84) and other criteria (HR=0.64; 95% CI, 0.52–0.78); and longer follow-up durations (⩾60 months; HR=0.66; 95% CI, 0.54–0.82) and shorter follow-up durations (<60 months; HR=0.54; 95% CI, 0.39–0.76).


Forest plots of the random-effect meta-analysis for the efficacy of tumour-infiltrating inflammatory cells for generalised tumour inflammatory infiltrate (A) and the CD3+ (B), CD8+ (C) and FoxP3+ (D) subsets stratified by infiltration location, including the tumour centre (CC), tumour stroma (ST) and invasive tumour margin (IM). The horizontal bars indicate the 95% CIs. The size of the square around each effect estimate indicates the weight of the individual study in the meta-analysis. Note: (A) Generalised tumour inflammatory infiltrate; (B1) CD3+ (CC); (B2) CD3+ (ST); (B3) CD3+ (IM); (C1) CD8+ (CC); (C2) CD8+ (ST); (C3) CD8+ (IM); (D1) FoxP3+ (CC); (D2) FoxP3+ (ST); (E) CCR7+.

Funnel plots of the relationship between the size of the effect in individual studies and the precision of the study estimate (log HR, horizontal axis; s.e., vertical axis) for generalised tumour inflammatory infiltrate (A) and T lymphocyte subgroups for OS (B).
CD3+ T lymphocyte subset
Seven studies (Figure 2B) investigated the association between the infiltration of CD3+ T lymphocytes and survival of CRC patients stratified by tumour location, with seven assessing the CC, three the ST and two the IM.
Tumour centre
Pooled estimates of the HR and 95% CI from the CC panel were provided for OS (HR=0.59; 95% CI, 0.31–1.12). Statistically significant heterogeneity was observed between studies (I2=82.5%, P<0.001). The estimated pooled HRs for CS and DFS were 0.89 (95% CI, 0.44–1.82) and 0.48 (95% CI, 0.35–0.66), respectively. Significant heterogeneity was observed for CS (I2=81.7%, P=0.02), whereas a lower level of heterogeneity was observed for DFS (I2=17.5%, P=0.30).
Tumour stroma
Three studies in each outcome panel investigated CD3+ infiltration in the ST; the pooled HRs of these three studies for OS and DFS were 1.20 (95% CI, 0.76–1.89) and 0.72 (95% CI, 0.24–2.10), respectively. However, these results should be interpreted with caution because of the small number of contributing studies and the significant evidence of heterogeneity between studies (I2=65%, P=0.06).
Invasive tumour margin
For the two studies investigating CD3+ infiltration at the IM, the pooled HRs for OS and DFS were 0.63 (95% CI, 0.42–0.93) and 0.48 (95% CI, 0.35–0.68), respectively. Although these results might indicate an association between improved survival and a high density of CD3+ infiltration, they should be interpreted with caution because of the small number of contributing studies.
CD8+ T lymphocyte subset
Eight studies provided data concerning the association between CD8+ infiltration and survival outcomes (Figure 2C), with seven assessing the CC, three the ST and three the IM.
Tumour centre
Pooled estimates of the HR and 95% CI from the CC panel were provided for OS (HR=0.67; 95% CI, 0.45–1.00). Statistically significant heterogeneity was observed between studies (I2=57.9%, P=0.05); however, no clear asymmetry was identified in the funnel plot (Figure 3B). The pooled HRs for CS and DFS were 0.64 (95% CI, 0.46–0.91) and 0.41 (95% CI, 0.20–0.82), respectively. Significant heterogeneity was observed for CS (I2=76.9%, P=0.013), whereas was no heterogeneity was observed for DFS (I2=0%, P=0.43).
Tumour stroma and invasive tumour margin
Three studies examined CD8+ infiltration in the ST, and a positive pooled effect was obtained for OS (HR=0.78; 95% CI, 0.67–0.92, n=3, I2=38%, P=0.38). Pooled estimates of two studies revealed a positive effect for OS (HR=0.91; 95% CI, 0.85–0.99) and DFS (HR=0.61; 95% CI, 0.37–1.00).
FoxP3+ Treg subset
Eight eligible studies provided estimates of the HR and 95% CI for the correlation between FOXP3+ Tregs and CRC survival (Figure 2D), with seven studies considering the CC and four considering the ST. For studies investigating the CC, the pooled HRs for OS, CS and DFS were 0.82 (95% CI, 0.58–1.17), 0.68 (95% CI, 0.52–0.90) and 0.45 (95% CI, 0.04–4.69), respectively. Moderate heterogeneity was noted in both the OS and DFS panels (OS: I2=44.40%, P=0.15; CS: I2=39.30%, P=0.19, respectively). The funnel plot for OS indicated that the HRs were symmetrically distributed, indicating a low risk of publication bias (Figure 3B). Significant heterogeneity was observed for DFS (I2=80.50%, P=0.002). Pooled analysis of studies investigating FOXP3+ Tregs in the ST did not indicate a prognostic impact regarding OS (HR, 0.54; 95% CI, 0.28–1.03; n=3, I2=57.60%) or DFS (HR, 0.48; 95% CI, 0.21–1.07; n=3, I2=59.10%).
CCR7+ subset
Three studies examined the association between CCR7+ infiltration and CRC survival (Figure 2E). The pooled HRs for OS and DFS were 0.51 (95% CI, 0.30–0.89) and 0.54 (95% CI, 0.28–1.03), respectively. There was no evidence of heterogeneity for OS (I2=0.0%, P=0. 537).
Other subsets
Only a small number of studies evaluating the prognostic impact of T lymphocyte subsets on CRC survival included CD45RO+ and GRB+. Therefore, we do not present the detailed pooled effects of these subsets even though the subgroup analysis revealed a favourable prognosis (summarised in Table 2).
Discussion
Our meta-analysis based on patient prognostic data from prospective and retrospective studies comparing high and low tumour inflammatory infiltrate densities in patients with CRC indicated that high generalised tumour inflammatory infiltrate alone could be a relatively pronounced predictive marker, with better associated outcomes than low infiltrate densities in terms of OS, CS and DFS. Similar findings favouring high infiltrate densities were provided in each individual study. The sensitivity analyses indicated the robustness of the HR estimates. We found no evidence of publication bias in the survival panels.
In contrast, the summary HRs across studies calculated for each subset of T lymphocytes stratified by infiltration location were not strong prognostic markers for CRC, with several of the pooled HRs moving close to or crossing one, indicating inconsistent findings of our available data. For example, in the original studies concerning CD3+ infiltration in CC, Galon et al (2006) and Guidoboni et al (2001) reported a favourable OS, with estimated HRs of 0.5 (95% CI, 0.37–0.66) and 0.4 (95% CI, 0.19–0.85), respectively. However, Deschoolmeester et al (2010, Lee et al (2010) and Nosho et al (2010) found that high intratumoural CD3+ infiltrate density was not associated with improved OS in CRC. Our meta-analysis does not support an association between high intratumoural CD3+ infiltration and OS (HR, 0.59; 95% CI, 0.31–1.12). Similarly, no significant association was observed for panels of cells other than T lymphocytes.
Inflammatory infiltration composed of lymphocytes is a common feature found in neoplasms. Nascent tumour cells are eliminated by the host innate and adaptive immune systems before the formation of a detectable tumour, a process that we call tumour immunosurveillance (Dunn et al, 2002). During the tumour-specific adaptive immune responses, some crucial components, such as cytotoxic T lymphocytes, can induce the production of tumour-associated antigen and the cytokine IFN-γ. IFN-γ has a pivotal role in antitumour immunity by inducing cell cycle arrest, differentiation, apoptosis, angiostasis and tumouricidal macrophage activity (Dunn et al, 2004). The great immune selection pressure in the host allows for the emergence of tumour cell variants that are able to avoid immune system attacks. The dual host-protective and tumour-promoting roles of immune cells are referred to as tumour immunoediting (Schreiber et al, 2011). The effects of the host immune response on tumour cell proliferation, survival, invasion, recurrence and metastasis are demonstrated by an analysis of in situ immunity. Studies have confirmed that immune infiltrates differ between tumour types and between individual patients. A variety of immune cell types could exist within one tumour, including innate and adaptive immune cells, which can be found at the CC, IM or ST or in the adjacent tertiary lymphoid structures, constituting what we have termed the immune contexture (Fridman et al, 2012). The immune contexture includes the type, density and location of adaptive immune cells, namely, CD3+, CD8+, FOXP3+ and CD45RO+ T cells. The results of several studies have indicated an association between tumour immune infiltrates and favourable disease outcomes, including survival, recurrence and metastasis (Nakano et al, 2001; Zhang et al, 2003; Sato et al, 2005; Galon et al, 2006; Pagès et al, 2009).
Subsets of immune cells are distributed differently among different tumour types, with the CC and IM being the most frequently involved areas. Different tumour types are affected by subsets of immune cells to different extents. Immune cell infiltration, particularly T lymphocyte infiltration, has been investigated in various tumour types and displays a strong correlation with improved outcome in breast, colorectal, prostatic, ovarian, biliary tract and other types of cancer (Sato et al, 2005; Galon et al, 2006; Flammiger et al, 2012; Goeppert et al, 2013; Seo et al, 2013).
To the best of our knowledge, this study is the first to systematically evaluate the prognostic effect of tumour-infiltrating inflammation stratified by lymphocyte subsets and infiltration site. The previous meta-analyses have not specifically focused on a certain cancer type and lack evidence of survival benefits in certain T lymphocyte subsets (Gooden et al, 2011; Kost et al, 2012). One strength of our study was the broad search strategy for articles that encompassed the most frequently used human T lymphocyte markers for CRC survival prediction and were published over the last 15 years. Thus, we were able to estimate the most appropriate prognostic markers for future clinical use.
However, these findings should be interpreted within the limitations of a meta-analysis based on observational studies because data are liable to be confounded by various factors, as reflected by the levels of heterogeneity. In addition, if studies met inclusion criteria, we did not further exclude them because of low methodological quality. In addition, we analysed potential confounders by performing a subgroup analysis. However, because of the small number of studies in each subgroup panel, the results of this analysis should be interpreted with caution. We assumed the following potential sources of bias. First, different cell scoring methods resulted in bias regarding the assignment of high and low lymphocyte infiltration. In our meta-analysis, cutoff points were used in several manners, with some studies choosing present/absent or few/extensive (Nagtegaal et al, 2001; Cianchi et al, 2002; Gao et al, 2005; Szynglarewicz et al, 2007), whereas other studies used the mean, median or quartiles (Naito et al, 1998; Guidoboni et al, 2001; Chiba et al, 2004; Menon et al, 2004; Galon et al, 2006; Salama et al, 2009; Frey et al, 2010; Lee et al, 2010; Nosho et al, 2010; Yoon et al, 2012) and related statistics; such differences might be responsible for the variability in reaching a standard threshold of a certain lymphocyte count. Second, although we performed subgroup analysis pertaining to prognostic associations between tumour location and T lymphocyte subtype, the tumour microenvironment was recognised as a complicated state of tumour–host interactions modulated by multiple cell societies inhabiting the epithelium and stromal unit. This environment consists of cytokines, chemokines, stromal cells and other factors in addition to lymphocytes (Liotta and Kohn, 2001; Li et al, 2007), which could result in specific tumour microenvironments of tumour-infiltrating inflammation and immune tolerance. Moreover, the type of immune cells or specific lymphocytes may be partial confounding factors. Other possible factors, such as tumour stage and microsatellite instability, also affect the prognosis of patients with CRC (Guidoboni et al, 2001; Menon et al, 2004; Nosho et al, 2010). However, those potential confounding variables varied considerably among individuals and thus yielded inconsistent prognostic results; multivariate analysis was performed in most of the included studies (n=23) to obtain more precise estimates, adjusting for clinicopathological variables. Third, for studies concerning T lymphocyte subgroups, some studies combined multiple markers and indicated that this combination of markers was a more sensitive predictor for recurrence and survival than a single T lymphocyte type, which was identified in intratumoural CD8+/FOXP3+ and CD3+/FoxP3+ cell ratios (Sinicrope et al, 2009; Suzuki et al, 2010). However, this finding must be investigated further because of the limited number of studies. Fourth, some HRs were derived from Kaplan–Meier survival curves when not provided by the original studies directly. To minimise this type of bias, we attempted to contact the authors for original data; furthermore, data were abstracted by two independent reviewers and cross-checked. However, the results should be still interpreted with caution because we may have failed to identify some published and unpublished studies with negative results that might have affected our pooled estimates. Although the funnel plots (Figure 3B) did not provide evidence of publication bias for OS stratified by T lymphocyte subset, we recognise that the use of relatively few studies might have decreased the power for detecting publication bias.
In summary, despite the above limitations, our findings suggest that generalised tumour inflammatory infiltration might serve as a robust marker for predicting the prognosis of patients with CRC. However, our findings do not provide strong evidence that a high density of T lymphocyte subsets in patients with CRC is a marker for good prognosis. With the establishment of improved and standardised evaluation protocols, a comprehensive assessment of tumour inflammatory infiltration might provide more valuable prognostic information for patients with CRC in the future. Future studies should use a prospective study design to improve the quality of clinical research and should consider the clinic-pathological variables of the patient, such as age, tumour location, tumour stage, KRAS mutation status, microsatellite instability and other tumour microenvironment factors. A strict follow-up scheme should also be used in future studies.
Change history
18 March 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Benson AB 3rd, Bekaii-Saab T, Chan E, Chen YJ, Choti MA, Cooper HS, Engstrom PF, Enzinger PC, Fakih MG, Fenton MJ, Fuchs CS, Grem JL, Hunt S, Kamel A, Leong LA, Lin E, May KS, Mulcahy MF, Murphy K, Rohren E, Ryan DP, Saltz L, Sharma S, Shibata D, Skibber JM, Small W Jr, Sofocleous CT, Venook AP, Willett CG, Gregory KM, Freedman-Cass DA (2013) Localized colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw 11 (5): 519–528.
Borenstein M, Hedges LV, Higgins JP, Rothstein HR (2011) Introduction to meta-analysis. Wiley: Chichester, UK.
Buckowitz A, Knaebel HP, Benner A, Blaker H, Gebert J, Kienle P, von Knebel Doeberitz M, Kloor M (2005) Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br J Cancer 92 (9): 1746–1753.
Chew A, Salama P, Robbshaw A, Klopcic B, Zeps N, Platell C, Lawrance IC (2011) SPARC, FOXP3, CD8 and CD45 correlation with disease recurrence and long-term disease-free survival in colorectal cancer. PLoS One 6 (7): e22047.
Chiba T, Ohtani H, Mizoi T, Naito Y, Sato E, Nagura H, Ohuchi A, Ohuchi K, Shiiba K, Kurokawa Y, Satomi S (2004) Intraepithelial CD8+ T-cell-count becomes a prognostic factor after a longer follow-up period in human colorectal carcinoma: possible association with suppression of micrometastasis. Br J Cancer 91 (9): 1711–1717.
Cianchi F, Palomba A, Messerini L, Boddi V, Asirelli G, Perigli G, Bechi P, Taddei A, Pucciani F, Cortesini C (2002) Tumor angiogenesis in lymph node-negative rectal cancer: correlation with clinicopathological parameters and prognosis. Ann Surg Oncol 9 (1): 20–26.
Correale P, Rotundo MS, Botta C, Del Vecchio MT, Ginanneschi C, Licchetta A, Conca R, Apollinari S, De Luca F, Tassone P (2012) Tumor infiltration by T lymphocytes expressing chemokine receptor 7 (CCR7) is predictive of favorable outcome in patients with advanced colorectal carcinoma. Clin Cancer Res 18 (3): 850–857.
Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, Tsang KY, Licchetta A, Mannucci S, Loiacono L, Tassone P, Francini G, Tagliaferri P (2010) Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother 33 (4): 435–441.
de Graeff P, Crijns AP, de Jong S, Boezen M, Post WJ, de Vries EG, van der Zee AG, de Bock GH (2009) Modest effect of p53, EGFR and HER-2/neu on prognosis in epithelial ovarian cancer: a meta-analysis. Br J Cancer 101 (1): 149–159.
Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, Vermorken JB (2010) Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol 11: 19.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3 (11): 991–998.
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22: 329–360.
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315 (7109): 629–634.
Flammiger A, Bayer F, Cirugeda-Kühnert A, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T (2012) Intratumoral T but not B lymphocytes are related to clinical outcome in prostate cancer. APMIS 120 (11): 901–908.
Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, Oertli D, Kettelhack C, Terracciano L, Tornillo L (2010) High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer 126 (11): 2635–2643.
Fridman WH, Pagès F, Sautès-Fridman C, Galon J (2012) The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12 (4): 298–306.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313 (5795): 1960–1964.
Gao JF, Arbman G, Wadhra TI, Zhang H, Sun XF (2005) Relationships of tumor inflammatory infiltration and necrosis with microsatellite instability in colorectal cancers. World J Gastroenterol 11 (14): 2179–2183.
Goeppert B, Frauenschuh L, Zucknick M, Stenzinger A, Andrulis M, Klauschen F, Joehrens K, Warth A, Renner M, Mehrabi A (2013) Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br J Cancer 109 (10): 2665–2674.
Gooden M, De Bock G, Leffers N, Daemen T, Nijman H (2011) The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer 105 (1): 93–103.
Guidoboni M, Gafa R, Viel A, Doglioni C, Russo A, Santini A, Del Tin L, Macri E, Lanza G, Boiocchi M, Dolcetti R (2001) Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathol 159 (1): 297–304.
Günther K, Leier J, Henning G, Dimmler A, Weißbach R, Hohenberger W, Förster R (2005) Prediction of lymph node metastasis in colorectal carcinoma by expressionof chemokine receptor CCR7. Int J Cancer 116 (5): 726–733.
Hayes DF, Bast RC, Desch CE, Fritsche H Jr, Kemeny NE, Jessup JM, Locker GY, Macdonald JS, Mennel RG, Norton L, Ravdin P, Taube S, Winn RJ (1996) Tumor marker utility grading system: a framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst 88 (20): 1456–1466.
Hedges LV, Olkin I, Statistiker M (1985) Statistical Methods for Meta-Analysis. Academic Press: New York.
Higgins JP, Thompson SG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21 (11): 1539–1558.
Huh JW, Lee JH, Kim HR (2012) Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch Surg 147 (4): 366–372.
Jass JR (1986) Lymphocytic infiltration and survival in rectal cancer. J Clin Pathol 39 (6): 585–589.
Kim M, Grimmig T, Grimm M, Lazariotou M, Meier E, Rosenwald A, Tsaur I, Blaheta R, Heemann U, Germer CT, Waaga-Gasser AM, Gasser M (2013) Expression of Foxp3 in colorectal cancer but not in Treg cells correlates with disease progression in patients with colorectal cancer. PloS One 8 (1): e53630.
Klintrup K, Makinen JM, Kauppila S, Vare PO, Melkko J, Tuominen H, Tuppurainen K, Makela J, Karttunen TJ, Makinen MJ (2005) Inflammation and prognosis in colorectal cancer. Eur J Cancer 41 (17): 2645–2654.
Kost SE, Kakal JA, Nelson BH (2012) The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res 18 (11): 3022–3029.
Lee WS, Park S, Lee WY, Yun SH, Chun HK (2010) Clinical impact of tumor-infiltrating lymphocytes for survival in stage II colon cancer. Cancer 116 (22): 5188–5199.
Li H, Fan X, Houghton J (2007) Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 101 (4): 805–815.
Liotta LA, Kohn EC (2001) The microenvironment of the tumour-host interface. Nature 411 (6835): 375–379.
Liska V, Daun O, Novak P, Treska V, Vycital O, Bruha J, Pitule P, Holubec L (2011) Tumor infiltrating lymphocytes as prognostic factor of early recurrence and poor prognosis of colorectal cancer after radical surgical treatment. Anticancer Res 31: 1990–1991. doi:10.5772/27244. Available from: http://www.intechopen.com/books/colorectal-cancer-biology-from-genes-to-tumor/tumor-infiltrating-lymphocytes-as-prognostic-factor-of-early-recurrence-and-poor-prognosis-of-colore.
Liu S, Lachapelle J, Leung S, Gao D, Foulkes WD, Nielsen TO (2012) CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res 14 (2): R48.
Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR (2011) Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 29 (15): 1949–1955.
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM (2005) REporting recommendations for tumour MARKer prognostic studies (REMARK). Br J Cancer 93 (4): 387–391.
Menon AG, Janssen-van Rhijn CM, Morreau H, Putter H, Tollenaar RA, van de Velde CJ, Fleuren GJ, Kuppen PJ (2004) Immune system and prognosis in colorectal cancer: a detailed immunohistochemical analysis. Lab Invest 84 (4): 493–501.
Nagtegaal ID, Marijnen CA, Kranenbarg EK, Mulder-Stapel A, Hermans J, van de Velde CJ, van Krieken JH (2001) Local and distant recurrences in rectal cancer patients are predicted by the nonspecific immune response; specific immune response has only a systemic effect—a histopathological and immunohistochemical study. BMC Cancer 1: 7.
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H (1998) CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58 (16): 3491–3494.
Nakano O, Sato M, Naito Y, Suzuki K, Orikasa S, Aizawa M, Suzuki Y, Shintaku I, Nagura H, Ohtani H (2001) Proliferative activity of intratumoral CD8+ T-lymphocytes as a prognostic factor in human renal cell carcinoma clinicopathologic demonstration of antitumor immunity. Cancer Res 61 (13): 5132–5136.
Nielsen HJ, Hansen U, Christensen IJ, Reimert CM, Brunner N, Moesgaard F (1999) Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. J Pathol 189 (4): 487–495.
Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, Giovannucci E, Dranoff G, Fuchs CS, Ogino S (2010) Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol 222 (4): 350–366.
Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, Glickman JN, Ferrone CR, Mino-Kenudson M, Tanaka N, Dranoff G, Giovannucci EL, Fuchs CS (2009) Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res 15 (20): 6412–6420.
Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, Lagorce C, Wind P, Marliot F, Bruneval P (2009) In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol 27 (35): 5944–5951.
Parmar MK, Torri V, Stewart L (1998) Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med 17 (24): 2815–2834.
Richards CH, Roxburgh CS, Anderson JH, McKee RF, Foulis AK, Horgan PG, McMillan DC (2012) Prognostic value of tumour necrosis and host inflammatory responses in colorectal cancer. Br J Surg 99 (2): 287–294.
Ropponen KM, Eskelinen MJ, Lipponen PK, Alhava E, Kosma VM (1997) Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer. J Pathol 182 (3): 318–324.
Roxburgh CS, Salmond JM, Horgan PG, Oien KA, McMillan DC (2009) The relationship between the local and systemic inflammatory responses and survival in patients undergoing curative surgery for colon and rectal cancers. J Gastrointest Surg 13 (11): 2011–2018, discussion 2018-2019.
Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B (2009) Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 27 (2): 186–192.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen YT, Ohtani H, Old LJ, Odunsi K (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102 (51): 18538–18543.
Schimanski CC, Schwald S, Simiantonaki N, Jayasinghe C, Gönner U, Wilsberg V, Junginger T, Berger MR, Galle PR, Moehler M (2005) Effect of chemokine receptors CXCR4 and CCR7 on the metastatic behavior of human colorectal cancer. Clin Cancer Res 11 (5): 1743–1750.
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331 (6024): 1565–1570.
Seo AN, Lee HJ, Kim EJ, Kim HJ, Jang MH, Lee HE, Kim YJ, Kim JH, Park SY (2013) Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br J Cancer 109 (10): 2705–2713.
Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG (2010) Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 59 (7): 926–933.
Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ (2009) Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology 137 (4): 1270–1279.
Suzuki H, Chikazawa N, Tasaka T, Wada J, Yamasaki A, Kitaura Y, Sozaki M, Tanaka M, Onishi H, Morisaki T, Katano M (2010) Intratumoral CD8(+) T/FOXP3 (+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol Immunother 59 (5): 653–661.
Szynglarewicz B, Matkowski R, Suder E, Sydor D, Forgacz J, Pudelko M, Grzebieniak Z (2007) Predictive value of lymphocytic infiltration and character of invasive margin following total mesorectal excision with sphincter preservation for the high-risk carcinoma of the rectum. Adv Med Sci 52: 159–163.
Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR (2007) Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 8: 16.
Tomsova M, Melichar B, Sedlakova I, Steiner I (2008) Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol Oncol 108 (2): 415–420.
Yoon HH, Orrock JM, Foster NR, Sargent DJ, Smyrk TC, Sinicrope FA (2012) Prognostic impact of FoxP3+ regulatory T cells in relation to CD8+ T lymphocyte density in human colon carcinomas. PloS One 7 (8): e42274.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN (2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348 (3): 203–213.
Zlobec I, Terracciano LM, Lugli A (2008) Local recurrence in mismatch repair-proficient colon cancer predicted by an infiltrative tumor border and lack of CD8+ tumor-infiltrating lymphocytes. Clin Cancer Res 14 (12): 3792–3797.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant no. 81372636) and the National High Technology Research and Development Program of China (863 program; grant no. SQ2014SFOZD00314).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Mei, Z., Liu, Y., Liu, C. et al. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer 110, 1595–1605 (2014). https://doi.org/10.1038/bjc.2014.46
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.46
Keywords
This article is cited by
-
A novel model for predicting the prognosis of postoperative intrahepatic cholangiocarcinoma patients
Scientific Reports (2023)
-
Tumour-infiltrating lymphocytes: from prognosis to treatment selection
British Journal of Cancer (2023)
-
Polyoxometalates based nanocomposites for bioapplications
Rare Metals (2023)
-
Usefulness of the preoperative inflammation-based prognostic score and the ratio of visceral fat area to psoas muscle area on predicting survival for surgically resected adenocarcinoma of the esophagogastric junction
Esophagus (2023)
-
CTNNB1 mutations, TERT polymorphism and CD8+ cell densities in resected hepatocellular carcinoma are associated with longer time to recurrence
BMC Cancer (2022)
