
Abstract
Background:
Standard therapy for acute promyelocytic leukaemia (APL) includes retinoic acid (all-trans retinoic acid (ATRA)), which promotes differentiation of promyelocytic blasts. Although co-administration of arsenic trioxide (ATO) with ATRA has emerged as an effective option to treat APL, the molecular basis of this effect remains unclear.
Methods:
Four leukaemia cancer human models (HL60, THP-1, NBR4 and NBR4-R2 cells) were treated either with ATO alone or ATO plus ATRA. Cancer cell survival was monitored by trypan blue exclusion and DEVDase activity assays. Gene and protein expression changes were assessed by RT-PCR and western blot.
Results:
ATO induced an antioxidant response characterised by Nrf2 nuclear translocation and enhanced transcription of downstream target genes (that is, HO-1, NQO1, GCLM, ferritin). In cells exposed to ATO plus ATRA, the Nrf2 nuclear translocation was prevented and cytotoxicity was enhanced. HO-1 overexpression reversed partially the cytotoxicity by ATRA-ATO in HL60 cells. The inhibitory effects of ATRA on ATO-mediated responses were not observed in either the ATRA-resistant NB4-R2 cells or in NB4 cells pre-incubated with the RARα antagonist Ro-41-52-53.
Conclusions:
The augmented cytotoxicity observed in leukaemia cells following combined ATO-ATRA treatment is likely due to inhibition of Nrf2 activity, thus explaining the efficacy of combined ATO-ATRA treatment in the APL therapy.
Similar content being viewed by others


Main
Acute promyelocytic leukaemia (APL) is a subtype of acute myeloid leukaemia (AML), characterised by a reciprocal chromosomal translocation t(15;17)(q22;q21) that generates the genetic fusion between the tumour suppressor – promyelocytic leukaemia (PML) – and the retinoic acid receptor-α (RARα) genes (Vardiman et al, 2009; Saeed et al, 2011). The thereby resulting PML–RARα fusion protein blocks transcription of genes involved in the differentiation programme of myeloid progenitors (Vardiman et al, 2009; Kamimura et al, 2011; Saeed et al, 2011). Currently, standard APL therapy includes retinoic acid, which promotes cell growth arrest, granulocytic differentiation and apoptosis of myeloid blasts (Huang et al, 1998) that are ascribed in part to augmented degradation of PML–RARα (Ablain et al, 2013) and increased RARα expression (Kamimura et al, 2011). Hence, standard chemotherapeutic regimens to treat APL involve the co-administration of all-trans retinoic acid (ATRA) together with anthracyclins (daunorubicin or idarubicin) or in combination with cytosine β-D-arabinofuranoside (Ara-C; Tallman and Altman, 2009; Kamimura et al, 2011). Despite the high rates of remission, undesirable outcomes are often observed after prolonged exposition to ATRA and chemotherapeutic drugs, such as the retinoic acid syndrome (Patatanian and Thompson, 2008), elevated mortality rates and emerging drug resistance (Kamimura et al, 2011). To avoid the development of such side effects, improved strategies are required during induction/consolidation therapies.
In addition to conventional therapies, administration of arsenic trioxide (ATO) alone (Chendamarai et al, 2012) or in combination with either retinoic acid or chemotherapeutic drugs (Iland et al, 2012; Park, 2012) has emerged as an effective option to treat newly diagnosed and relapsed APL patients (Shen et al, 1997; Chendamarai et al, 2012; Iland et al, 2012), which results in lower early mortality rates and higher percentages of complete remission and disease-free survival (Tallman and Altman, 2009; Kamimura et al, 2011). Furthermore, ATO treatment has been shown to be effective against other AML subtypes (Wetzler et al, 2011), myelodisplastic syndrome (Sekeres et al, 2011) and multiple myeloma (Sharma et al, 2012). ATO effectiveness in APL patients may be explained not only by its general cytotoxic effect, but also by its ability to promote PML–RARα degradation (Zhang et al, 2010; Lang et al, 2012). Despite these promising results, acquired resistance is observed in patients treated with ATO-ATRA (Goto et al, 2011). Retinoic acid together with ATO is known to enhance PML–RAR degradation. However, whether ATRA alters cellular responses triggered by ATO and inhibits survival pathways required for the development of ATO resistance remains to be determined.
Survival of leukaemia cells following treatment with chemotherapeutic drugs is associated with activation of the Nrf2 pathway (Rushworth and MacEwan, 2011). Under normal conditions, cytosolic Nrf2 levels are maintained low by the physical interaction with Keap1, which promotes Nrf2 ubiquitination and subsequent proteasome-mediated degradation (Ma, 2012). In response to electrophilic or oxidative stress, S-alkylation or oxidation of cysteine residues in Keap1 releases Nrf2 from this complex and permits translocation to the nucleus where Nrf2 recognises the enhancer antioxidant/electrophilic response elements present in the promoter of cytoprotective and detoxifying genes. Examples of such genes include the phase-I drug oxidation enzyme NAD(P)H: quinone oxidoreductase 1 (NQO1), the cytoprotective haem-oxygenase-1 (HO-1) and the antioxidant enzymes glutamyl cysteine ligase modulator (GCLM) and -catalytic subunits, involved in GSH synthesis, ferritin among others (Rushworth and MacEwan, 2011). Notably, an additional mechanism regulating Nrf2 activity has been identified in breast cancer cells, where Nrf2 transcriptional activity is inhibited by the physical interaction with RARα in the presence of ATRA (Wang et al, 2007, 2013). At present, it is not known whether a similar inhibitory mechanism operates in AML cells and, particularly, in the APL subtype.
In this study, we provide evidence showing that inhibition by ATRA of the Nrf2-dependent antioxidant response triggered by ATO correlates with enhanced cytotoxicity in NB4 APL cells. This effect was dependent on RARα function because Nrf2 activity was not affected by ATRA in the ATRA-resistant NB4-R2 cells and RARα antagonist Ro-41-5253 precluded ATRA-mediated inhibition in NB4 parental cells.
Materials and Methods
Drugs and chemicals
Zinc (II) protoporphyrin IX (ZnPP) and Ro-41-5253 were purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA); copper (II) protoporphyrin IX (CuPP) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA); ATRA, Ara-C, daunorubicin, methotrexate, tert-butylhydroquinone (t-BHQ), L-buthionine-sulfoximine (BSO) and ATO were from Sigma-Aldrich (St Louis, MO, USA).
Cell lines and culture conditions
The AML-derived cell lines HL60 and THP-1 were purchased from the European Cell Culture Collection (Salisbury, UK). The APL-derived NB4 and NB4-R2 cells, a subline derived from NB4 cells that bears a missense mutation in the PML fragment of PML–RARα, which inhibits RARα transcriptional function (Duprez et al, 2000), were kindly gifted by Mario P Tshan (University of Bern, Bern, Switzerland). Cells were cultured in RPMI 1640 medium (Gibco, Paisley, UK) supplemented with 2 mM glutamine, 10% fetal bovine serum (Hyclone, Logan, UT, USA) and antibiotics (100 U ml−1 penicillin, 100 μg ml−1 streptomycin) in a humidified atmosphere with 5% CO2 at 37 °C.
Western blot analysis
Total-cell extracts were prepared in lysis buffer (Triton 1%, leupeptin 12.5 μg ml−1, antipain 10 μg ml−1, benzamidine 100 μg ml−1, phenylmethanesulphonylfluoride 1 mmol l−1 and sodium orthovanadate 1 mmol l−1) and protein concentrations were determined using the Bio-Rad Protein Assay reagent following the manufacturer’s instructions (Bio-Rad, Hertfordshire, UK). Total-protein extracts (40 μg per lane) were separated by SDS–PAGE in 10% mini-gels (Bio-Rad) and transferred to nitrocellulose as described (Valenzuela et al, 2010). Blots were blocked with 5% milk in TBS-Tween-20 and then probed with different antibodies. Mouse monoclonal anti-β-actin (using a dilution of 1 : 30 000) and anti-HO-1 (1 : 1000) were purchased from Abcam (Cambridge, UK), whereas anti-NQO1 (1 : 1000), rabbit polyclonal anti-Nrf2 (1 : 1000), anti-TBP1 (1 : 1000) and anti-HSP90 (1 : 3000) were obtained from Santa Cruz Biotechnology. Bound first antibodies were detected with horseradish peroxidase-conjugated anti-mouse (1 : 2500, Dako, Glostrup, Denmark) or anti-rabbit (1 : 2500, Millipore, Billerica, MA, USA) secondary antibodies followed by incubation with the Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, St Leon-Rot, Germany) and exposure to BioMax MR film (Kodak, Brussels, Belgium).
Analysis of mRNA levels
Total RNA was isolated using the Purezol RNA isolation reagent following instructions provided by the manufacturer (Bio-Rad). RNA samples were employed as templates to generate total complementary DNA (cDNA) using the M-MLV reverse transcriptase (Promega, Madison, WI, USA) and the oligo (dT) 15 primers (Promega).
cDNAs were amplified by PCR with the following specific primers:
HO-1 (sense primer 5′-CAAAGTGCAAGATTCTGCCC-3′; antisense primer 5′-CACATGGCATAAAGCCCTAC-3′),
NQO1 (sense primer 5′-CAAATCCTGGAAGGATGGAA-3′; antisense primer 5′-AAGTGATGGCCCACAGAAAG-3′),
GCLM (sense primer 5′-AGCGAGGAGCTTCATGATTG-3′; antisense primer 5′-GAACAGGCCATGTCAACTGC-3′),
ferritin heavy chain (sense primer 5′-CTGGAGCTCTACGCCTCCTA-3′; antisense 5′-TGGTTCTGCAGCTTCATCAG-3′) and
GADPH (sense primer 5′-TCAACGACCACTTTGTCAAGC-3′; antisense primer 5′-CCAGGGGTCTTACTCCTTGG-3′).
All amplicons were obtained after 30 amplification cycles, each of which involved consecutive 1-min steps at 94, 58 and 72 °C and visualised by electrophoresis in agarose gels (2%).
Trypan blue exclusion assay
Cells were plated at a density of 3.5 × 105 per well and incubated for 48 h in the presence of drugs. Cell viability was evaluated by the trypan blue exclusion assay with the automated cell counter TC10 (Bio-Rad). The loss of membrane integrity can be measured by using exclusion dyes that cannot enter living healthy cells but that are taken up by dying cells with permeabilized plasma membranes. Although this assay does not discriminate between different forms of cell death, the loss of plasma membrane integrity is considered a point of no return, while the occurrence of other biochemical events does not necessarily mean that cell death will ensue (Kroemer et al, 2009).
DEVDase activity
Caspase activity in cell lysates was determined by quantifying the release of the fluorescent dye 7-amino-4-trifluoromethylcoumarin (AFC) from the caspase-3 substrate Asp-Glu-Val-Asp-AFC (Enzo Life Sciences) in the multilabel plate reader Victor X2 (Perkin Elmer, Waltham, MA, USA). A unit of enzymatic activity was defined as 1 μmol of substrate transformed min−1 per mg protein extract (Nicholson et al, 1995).The general caspase inhibitor Q-VD-OPh (R&D Systems, Oxfordshire, UK) was added at 10 μ M in the reaction as a control.
GSH content
Total GSH content was evaluated following formation of the yellow derivative 5′-thio-2-nitrobenzoic acid (TNB) produced during the reaction of GSH with the sulphydryl reagent 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) at 412 nm. GSSG was determined in a similar manner, after removal of GSH with 2-vinylpyridine as described (Rahman et al, 2006). GSH depletion (90%) was achieved by treating cells for 24 h with BSO (10 μ M).
Nuclear/cytoplasmic protein extraction
Cells were collected, washed in PBS pH 7.2 and re-suspended in a hypotonic buffer (Hepes-KOH 10 mM, MgCl2 2 mM, EDTA 0.1 mM, KCl 10 mM, NP40 0.5%, DTT 1 mM, PMSF 0.5 mM, pH 7.9) for 10 min on ice. Lysates were centrifuged at 13 000 g for 1 min. Supernatants (cytoplasmic fractions) were recovered and pellets were re-suspended in a hypertonic buffer (Hepes-KOH 50 mM, MgCl2 2 mM, EDTA 0.1 mM, NaCl 400 mM, KCl 50 mM, glycerol 10%, DTT 1 mM, PMSF 0.5 mM, pH 7.9). Nuclei were allowed to swell on ice for 30 min. After centrifugation (13 000 g for 15 min), the supernatants were recovered and analysed by immunoblotting.
Constructs
The plasmid pSFFV-HO-1 was constructed by cloning the human HO-1 cDNA obtained by PCR using the sense primer 5′-CCGGAATTCGGGATGGAGCGTCCGCAACCC-3′ and antisense 5′-CCGGAATTCTCACATGGCATAAAGCCCTA-3′ primers in the EcoRI restriction site of the pSFFV-Neo plasmid (plasmid 8769; Addgene, Cambridge, MA, USA). The short hairpin RNA (shRNA) expression vector was obtained by cloning a shRNA cassette directed against human Nrf2 mRNA (shRNA duplex sense 5′-GATCCGTAAGAAGCCAGATGTTAAGTCAAGAGCTTAACATCTGGCTTCTTACTTTTTTTTGGAA-3′ and antisense 5′-AGCTTTCCAAAAAAAAGTAAGAAGCCAGATGTTAAGCTCTTGACTTAACATCTGGCTTCTTACG-3′) in the BamHI/HindIII cloning sites of the pRS vector (TR2003, Origene, Rockville, MA, USA). A plasmid containing a non-effective 29-mer scrambled shRNA cassette (TR30012, Origene) was used as control. Molecular cloning was performed with material purchased from Fermentas (St Leon-Rot, Germany) and Favorgene (Viena, Austria) (DNA purification kits).
Transfection/electroporation
HL60 cells were electroporated using the 4D-Amaxa Nucleofector kit (Lonza, Verviens, Belgium) following instructions provided by the manufacturer. For stable transfection, cells were serially diluted and clones were selected in the presence of G418 (1000 μg ml−1) for 10 days.
Differentiation assay
Differentiation of cells was assessed by the nitroblue tetrazolium (NTB) reduction assay following 3 days of ATRA treatment (Gupta et al, 2011). Briefly, myelocytic blasts (that is, HL60, THP-1, NB4) respond to ATRA undergoing a terminal granulocytic maturation and losing their ability to proliferate. These differentiated promyelocytes also acquire the ability to trigger a respiratory burst. Thus, when they are pretreated with 100 ng ml−1 phorbol myristate acetate, the superoxide anion generated during the respiratory burst reacts with NTB to produce a blue–purple precipitate.
Statistical analysis
All data are expressed as the mean±s.e.m. of at least three independent experiments. Data were processed using INSTAT v. 3.05 (GraphPad Software, San Diego, CA, USA, www.graphpad.com). Statistical significance of differences (P<0.05) was determined using the one-way analysis of variance test and the Dunnett or Tukey comparison post tests.
Results
Characterisation of ATO-mediated Nrf2 activation in AML cells
In both HL60 and THP-1 cells, a rapid Nrf2 nuclear accumulation was observed, which remained stable from 3 to 12 h and it decreased after 24 h incubation in the presence of 6.25 μ M ATO (Figure 1A). The concomitant increase in the expression of two downstream Nrf2 targets (NQO1 and HO-1) remained fairly stable up to 24 h incubation with ATO (6.25 μ M). No changes in Nrf2 expression were observed in untreated cells (basal conditions) during the 24 h incubation time (data not shown). This enhanced protein expression was associated with transcriptional activation of Nrf2 targets by ATO and by t-BHQ, a well-known electrophilic Nrf2 activator, as shown by RT-PCR analysis of HO-1, NQO1, GCLM and ferritin mRNA levels (Figure 1B, upper panels). Interestingly, nuclear accumulation of Nrf2 was more pronounced when cells were treated with ATO as compared with t-BHQ (Figure 1B, lower panels). To confirm that the observed induction of downstream targets was really dependent on Nrf2 activation, its expression was silenced by using shRNA (shNrf2 cells). In shNrf2 cells, induction of both HO-1 and NQO1 by ATO was reduced (Figure 1C).

ATO induced Nrf2 targets in AML cells. (A) HL60 and THP-1 nuclear (N) or total (T) protein levels of Nrf2, TBP1, HO-1, NQO1 and β-actin. Cells were incubated for 24 h with 6.25 μ M ATO. (B) Reverse-transcription PCR (upper panels) analysis of GAPDH, HO-1, NQO1, GCLM and ferritin targets mRNA levels and nuclear Nrf2 translocation (bottom panels) in HL60 and THP-1 cells following ATO (6.25 μ M) or t-BHQ (25 μ M) treatments for 24 h. (C) HL60 cells were electroporated either with the pRS-control or pRS-shNrf2 plasmids and treated with ATO (6.25 μ M) for 12 h. Total protein extracts were analysed for the presence of Nrf2, NQO1, HO-1 and β-actin by immunoblotting. (D) HL60 and THP-1 cells were treated with different concentrations of ATO, Ara-C or daunorubicin for 24 h, as indicated. Following treatment, total protein extracts were prepared and NQO1, HO-1 and β-actin protein levels were evaluated by immunoblotting. A typical western blot out of three experiments is shown. (E) GSH content was determined in HL60 and THP-1 cells treated with ATO (6.25 μ M), Ara-C (5 μ M), daunorubicin (0.5 μ M) or t-BHQ (25 μ M) for 24 h. Statistically significant differences with respect to the control condition are indicated (means±s.e.m.; n=3; *P⩽0.05, **P⩽0.01, ***P⩽0.001). Ctl, control.
To investigate whether the presence of chemotherapeutic drugs currently used in AML therapy was sufficient to modulate Nrf2 activation, both HL60 and THP-1 cells were exposed for 24 h to ATO (6.25–12.5 μ M), Ara-C (1–5 μ M) or daunorubicin (0.25–1 μ M). Western blot analysis revealed that exposure of cells to ATO induced NQO1 and HO-1 expression, but these proteins were not detected when cells were treated with Ara-C or daunorubicin (Figure 1D). Notably, the highest concentration of ATO (25 μ M) did not induce the expression of either NQO1 or HO-1, likely due to an inhibitory effect on transcription as a consequence of DNA damage, as recently reported (Nakamura et al, 2013). Regarding GSH levels, an elevated intracellular content was observed only when cells were exposed for 24 h to both ATO (6.25 μ M) and t-BHQ (25 μ M), whereas in the presence of either Ara-C (5 μ M) or daunorubicin (0.5 μ M) no GSH increase was observed (Figure 1E). These data indicate that ATO triggers in AML cells an antioxidant response characterised by Nrf2 activation and induction of its downstream targets.
The effects of ATO on both Nrf2 nuclear translocation and target induction were inhibited by N-acetyl-cysteine (NAC) but not by other antioxidant molecules like mannitol or tiron (Supplementary Figure 1A and B). In this context, the effect by NAC is in agreement with a previous report showing the use of thiol compounds as antidote against ATO overdoses (Isbister et al, 2004). Furthermore, no ROS formation was detected at low concentration of ATO in AML cells loaded with the fluorescent dye DCFH-DA (Supplementary Figure 1C). These results suggest that ROS is not the main mechanism triggering Nrf2 activation in cells treated with low ATO concentrations.
Pharmacologic inhibition of HO-1 or GSH depletion sensitises AML and APL cells to ATO
Antioxidant elements induced during the activation of Nrf2 pathway (that is, HO-1 and GSH) may have a critical role in a putative resistance against ATO-mediated cytotoxicity in both AML and APL cells. To explore this likelihood, HL60, THP-1 and the NB4 (PML–RARα/RARα) cells were first incubated with different concentrations of ATO in the presence or absence of either the active HO-1 inhibitor ZnPP or CuPP, its inactive form. As shown in Figure 2A, ZnPP (5 μ M) augmented cell cytotoxicity in HL60 and THP-1 exposed to ATO for 48 h, as determined by the trypan blue exclusion assay. This effect was not observed when CuPP (5 μ M) was used as a control (data not shown). Interestingly, NB4 cells were sensitive to lower ATO concentrations and the effect of ZnPP was limited to concentrations varying between 0.35 and 0.75 μ M ATO. To discard unspecific effects of ZnPP, K562 cells (which do not induce HO-1 in response to either ATO or t-BHQ) were subjected to similar conditions. ATO-mediated cytotoxicity was not affected by the presence of ZnPP in these cells (Supplementary Figure 2). Regarding the apoptosis-like induced cell death, the DEVDase assay indicated that caspase activity was increased by three-fold when HL60 cells were treated with 6.25 μ M ATO in the presence of 5 μ M ZnPP as compared with ATO alone (Figure 2B). Moreover, the incubation of HL60 cells in the presence of 5 μ M ZnPP did not modify the caspase activation induced by methotrexate, daunorubicin or Ara-C (Supplementary Figure 3).

Pharmacologic inhibition of Nrf2 targets (HO-1 and GSH) sensitises AML and APL cells to ATO. (A) Cell viability was determined in HL60, THP-1 and NB4 cells exposed for 48 h to different concentrations of ATO (0–25 μ M) in the presence (triangles) or absence (squares) of ZnPP (5 μ M) using the trypan blue exclusion assay. (B) HL60 cells treated with ATO (6.25 μ M) in the presence of ZnPP (5 μ M) for 24 h. Cell extracts were prepared and DEVDase activity was assessed as described in the Materials and Methods section. The pan-caspase inhibitor 10 μ M Q-VD-OPh (QVD) was included as a negative control (data not shown). (C–D) HL60 and NB4 cells were pre-incubated in the presence or the absence of BSO (10 μ M) for 24 h (+BSO) and then treated with ATO (6.25 and 0.75 μ M, respectively) for an additional 12 h. Cytotoxicity was determined either by (C) trypan blue exclusion assay or (D) DEVDase activity. Statistically significant differences with respect to the control condition are indicated (means±s.e.m.; n=3; *P⩽0.05, **P⩽0.01, ***P⩽0.001).
In a second step, the GSH content in HL60 and NB4 cells was modulated by pre-incubating with 10 μ M BSO, an inhibitor of GSH synthesis, which depletes its content by ∼90% (data not shown). In this context, the ATO-mediated cell death – when used at 6.25 μ M in HL60 cells and at 0.75 μ M in NB4 cells, respectively – was notably augmented, as determined by trypan blue exclusion assay (Figure 2C) and DEVDase activity (Figure 2D) after 12 h treatment.
These results indicated that interference with Nrf2-induced targets, namely, HO-1 expression and GSH levels, led to an increased ATO-mediated cytotoxicity. Consequently, these Nrf2-induced targets should have a protective role during ATO challenge in both AML and APL cells.
The association of ATRA and ATO inhibited Nrf2 nuclear translocation and leads to an enhanced cell death in both AML and APL cells
As ATRA inhibited the Nrf2 transcriptional activity in breast cancer cells (Wang et al, 2007), we explored whether retinoic acid may modulate the characterised Nrf2 activation in response to ATO in both AML and APL cells. Rather strikingly, when HL60 and NB4 cells were exposed to ATO in the presence of 1 μ M ATRA, Nrf2 nuclear translocation, induction of HO-1 and NQO1 expression (Figure 3A), as well as the increase in GSH content (Figure 3B) were reduced.

The association of ATRA and ATO inhibited Nrf2 nuclear translocation promoting cell death in both AML and APL cells. HL60 and NB4 cells were pretreated with ATRA (1 μ M) for 2 h and then exposed to ATO (3.1–6.25 μ M; 12 h and 0.75–1.5 μ M; 24 h), respectively. (A) After treatment, total and nuclear protein extracts were prepared and HO-1, NQO1, Nrf2 and β-actin protein levels were assessed by immunoblotting. Illustrations of typical western blots are shown. (B) GSH content in both HL60 and NB4 cells, under the same conditions described above, was determined as described in the Materials and Methods section. Cytotoxicity was assessed by using (C) trypan exclusion assay and (D) DEVDase activity. HL60 and NB4 cells were exposed to ATO (6.25 and 0.75 μ M) for 24 or 48 h, respectively. Statistically significant differences with respect to the control condition are indicated (means±s.e.m.; n=3; *P⩽0.05, **P⩽0.01, ***P⩽0.001). Ctl, control.
Interestingly, the ATRA-mediated inhibition of ATO-induced Nrf2 activity was accompanied with an enhanced cytotoxicity. Cancer cell viability, as determined by the trypan blue exclusion assay (Figure 3C), was 80 and 65% in HL60 and NB4 cells, respectively, when exposed to ATO alone. However, in cells pretreated for 2 h with ATRA (1 μ M) and then further incubated for 24 h with ATO, the cell viability was decreased to 20 and 40% in HL60 and NB4 cells, respectively. Similarly, the caspase activation as measured by using the DVEDase assay (Figure 3D) was enhanced from five-fold (ATO alone) to seven-fold when ATO was associated with ATRA in HL60 cells. In NB4 cells, the activation of caspase was enhanced from 3.5-fold (ATO alone) to 5.5-fold (ATO+ATRA).
Ectopic expression of HO-1 in HL60 cells precluded ATRA/ATO-promoted cytotoxicity
Previous results show that HO-1 expression – via Nrf2 activation – would have a major protective role in the survival of cells exposed to both ATRA and ATO. To explore this, HL60 cells were stably transfected with a plasmid expressing the human HO-1 gene (pSFFV-HO-1). As shown in Figure 4A, the SFFV promoter was activated in these cells and ectopic HO-1 expression was maintained even in the presence of ATRA-ATO combination. Such forced HO-1 expression was sufficient to reduce ATO-mediated cytotoxicity, but partially reverted (not significant) the enhanced cytotoxicity triggered by ATRA during ATO challenge, as determined by the trypan blue exclusion (Figure 4B) and DEVDase activity (Figure 4C) assays.

Ectopic expression of HO-1 in HL60 cells precluded ATRA-promoted cell death. (A) HL60 cells, stably transfected with either pSFFV-neo or pSFFV-HO-1, were incubated for 12 h with ATO (6.25 μ M) with or without ATRA (1 μ M). Total protein extracts were prepared and HO-1 protein levels were evaluated by immunoblotting. Illustration of a typical western blot is shown. Cytotoxicity following ATO and/or ATRA treatment was assessed by either (B) trypan blue exclusion or (C) DEVDase activity assays after 24 or 48 h of treatment, respectively. Statistically significant differences with respect to the control condition are indicated (means±s.e.m.; n=3; *P⩽0.05). Ctl, control.
Taken together, these results confirm that ATRA synergizes with ATO to enhance cytotoxicity by inhibiting the Nrf2 pathway in AML cells.
Involvement of RARα in ATRA-mediated Nrf2 inhibition
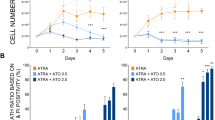
Previous results showed that ATRA enhances ATO-mediated cytotoxicity by inhibiting the Nrf2 pathway in AML cells. This led us to explore the potential role of RARα in the ATRA-mediated effects on Nrf2 pathway in response to ATO. To this end, in addition to NB4 cells, we included in our study the ATRA-resistant NB4-R2 cells, a cellular subline bearing a missense mutation in the PML fragment of PML–RARα that inhibits RARα transcriptional function (Duprez et al, 2000). These cells do not differentiate even following 3 days of exposure to 1 μ M ATRA (Figure 5A). Conversely, HL60 and NB4 were able to differentiate under such conditions. Subsequently, NB4-R2 and NB4 cells were incubated with ATO-ATRA. Western blot analysis revealed that HO-1 induction was not reduced in NB4-R2 cells treated with ATO in the presence of ATRA, as was the case for NB4 parental cells (Figure 5B). Similar results were observed when GSH content was determined (Figure 5C).

Loss of ATRA-mediated Nrf2 inhibitory mechanism in the NB4-R2 retinoic acid-resistant cell line. (A) HL60, NB4 and NB4-R2 cells were treated for 3 days with ATRA 1 μ M. Differentiation was determined by the NTB reduction assay as indicated in Materials and Methods. (B) NB4-R2 and NB4 cells were treated with ATO (0.75 μ M) in the presence of ATRA (1 μ M) for 24 h. After treatment, total protein extracts were prepared and HO-1 and β-actin protein levels were assessed by immunoblotting. Illustration of a typical western blot is shown. (C) GSH content was measured in NB4-R2 cells treated with ATO (0.75 μ M) in the presence of ATRA (1 μ M) for 24 h. Cell survival was assessed in NB4-R2 cells treated with ATO (0.75 μ M) in the presence of ATRA (1 μ M) for 24 h using (D) DEVDase activity or (E) the trypan blue exclusion assays. (F) NB4 cells were incubated with Ro-41-5351 (0.5 μ M) for 24 h and then treated with ATRA (1 μ M) and ATO (0.75 μ M) for additional 24 h. Cell extracts were collected and cell survival was assessed by using the trypan blue exclusion assay. Statistically significant differences with respect to the control condition are indicated (means±s.e.m.; n=3; *P⩽0.05, **P⩽0.01, ***P⩽0.001). Ctl, control.
The critical role of RARα and ATRA interaction in Nrf2 responses to ATO was further demonstrated by recording the cytotoxicity by ATO in both cell lines. As previously shown in NB4 cells (Figure 3C and D), the cytotoxicity by ATO alone was strongly enhanced by associating ATRA plus ATO. However, cytotoxicity was not augmented in NB4-R2 cells when treated with ATO in the presence of ATRA as determined by the trypan blue exclusion (Figure 5D) and DEVDase (Figure 5E) assays. To evaluate whether RARα activation by ATRA was indeed necessary to augment cytotoxicity, NB4 cells were pretreated with the RARα antagonist Ro-41-5253 (Apfel et al, 1992) and then incubated with ATO-ATRA for an additional 24 h. Interestingly, Ro-41-5253 eventually abolished the ATRA-mediated increase in cytotoxicity (Figure 5F).
Discussion
The present study was undertaken to contribute in the elucidation of the molecular mechanisms underlying the promising anti-cancer effects shown by ATRA-ATO, particularly in APL therapy. We characterised the Nrf2 activation in AML cells by analysing the induction of known Nrf2 targets in response to different drugs used currently in APL therapy, that is, Ara-C, daunorubicin and ATO. Results obtained showed that ATO was the only drug able to induce Nrf2 targets (HO-1, NQO1, GSH content). Although ATO-mediated cytotoxicity has been associated with ROS generation (Alarifi et al, 2013), Nrf2 activation by ATO in AML cells appeared to be independent of ROS formation. Indeed, in agreement with a previous study (Morales et al, 2009), antioxidants such as mannitol and tiron (a metal chelator and scavenger of both superoxide and hydroxyl radical species) were unable to preclude Nrf2 activation (Supplementary Figure 1A and B). In addition, a rapid activation of Nrf2 by ATO (1 μ M) via a noncanonical pathway, independent of ROS formation, has been recently reported (Lau et al, 2013). The authors reported that arsenic blocks authophagic flux, resulting in accumulation of p62 and sequestration of Keap1 into autophagosomes that leads to a prolonged Nrf2 activation. The mechanism by which ATO induces the formation of ROS has not been totally elucidated, but upregulation of NADPH oxidase and subsequent ROS production appear as one of the main intracellular target induced in NB4 cells treated for 10 days with low doses of ATO (Chou et al, 2004). Under our experimental conditions, the formation of ROS by ATO appears to be highly dependent on utilised doses. For instance, by using the fluorescein probe (DCFH-DA), the detection of ROS was only observed at ATO doses higher than 25 μ M, whereas at concentrations lower than 12 μ M no ROS formation was detected (Supplementary Figure 1C). These results are consistent with previous reports showing that concentrations of ATO higher than 12 μ M (Alarifi et al, 2013) or doses of sodium arsenite higher than 80 μ M (Chen et al, 1998) lead to the formation of ROS. In this context, the activation of Nrf2 pathway and further protection of cells against ATO-mediated cytotoxicity by lipoic acid (Wang et al, 2011) or NAC may be explained by covalent binding of ATO to sulphydryl residues in proteins or other thiol-rich molecules (Zhang et al, 2010). Accordingly, NAC inhibited Nrf2 nuclear translocation mediated by ATO (Supplementary Figure 1A).
HO-1, catalysing the catabolism of pro-oxidant haem to carbon monoxide, biliverdin and free iron (Gozzelino et al, 2010), has been shown to protect AML cells against standard chemotherapeutic drugs, proteasome inhibitors and TNF-α-induced apoptosis (Rushworth et al, 2010). Despite the fact that HO-1 induction is a biological marker for ATO exposition (Miller et al, 2002), there is controversy as to whether HO-1 protects cells against ATO-induced cytotoxicity (Morales et al, 2009; Wang et al, 2011). Our results underline the major role of HO-1 to resist the ATO-mediated cytotoxicity in AML cells. Although a previous study showed that HO-1 induction protected AML cells against daunorubicin and Ara-C (Heasman et al, 2011), our data demonstrated that it was not the case even when HO-1 was inhibited with ZnPP in HL60 cells (Supplementary Figure 3). In addition, GSH content is considered relevant in the resistance to ATO-mediated cytotoxicity and high GSH content had been related with a less sensitivity to ATO (Davison et al, 2003). Indeed, when GSH synthesis was inhibited with BSO, we observed an augmented DEVDase activity with a concomitant loss of cell viability. Taken together, these results support the involvement of both HO-1 and GSH in cell sensitivity to ATO.
ATRA-ATO co-treatment has shown to be an effective option in the APL therapy (Chendamarai et al, 2012); therefore, we investigated whether the characterised Nrf2 protective activity may be modified by ATRA during ATO challenge. As shown here, ATRA inhibited Nrf2 activity during ATO challenge and as expected, it was associated with enhanced cytotoxicity (Figure 3C and D). Notably, this inhibitory effect was not observed in the NB4-derived ATRA-resistant NB4-R2 cells (PML–RARα+, RARα+). These cells were previously characterised as containing a mutation in the RARα E-domain of PML–RARα that substitutes Gln903 for a stop codon, generating a truncated protein that displays a dominant negative effect on RARα transcriptional activity (Duprez et al, 2000). Results obtained in NB4-R2 cells (Figure 5) are similar to those in MCF-7 cells, where RARα expression was silenced by shRNA, resulting in the abolishment of ATRA-mediated inhibition of Nrf2 activity (Wang et al, 2007). To confirm a major role played by RARα, NB4 cells were pre-incubated with the RARα antagonist Ro-41-5153. As expected, a potentiating effect by ATRA on cytotoxicity was also precluded (Figure 5E). This led us to hypothesise that a physical interaction between RARα and Nrf2 may occur during ATO-ATRA treatment in AML cells.
In a clinical study conducted by Goto et al (2011), a relapsed APL patient following chemotherapy with insufficient response to ATRA was later treated with a combination protocol of ATO plus ATRA, and promyelocytes resistant to ATO were isolated during the terminal stage of its clinical course. Interestingly, a clonal expansion of two subpopulations of blasts was detected. A main population presented missense mutations in both the B2 and LBD domains of PML–RARα and a minor emergent population with a missense mutation in the LBD domain. To note, mutations in the retinoic acid-binding domain in PML–RARα are related with ATRA resistance (Marasca et al, 1999; Zhou et al, 2002). On the basis of these results, we would like to hypothesise that the presence of these mutations generating ATRA resistance may be linked to ATO resistance via the Nrf2 pathway. In this context, when the ATRA-resistant NB4-R2 and NB4 cells were treated with ATO, a stronger induction of HO-1 protein was observed in the former cells (Figure 5B). A link between ATRA and ATO resistance via the Nrf2 pathway may thus be suggested.
Total elimination of promyelocytic blasts during chemotherapy seems to be crucial in APL. Indeed, recent studies indicated that even being differentiated with retinoids, blasts harbouring the genetic fusion PML–RARα still conserved the capacity to reboot APL in an animal model (Ablain et al, 2013). Thus, molecular bases of resistance to ATO are necessary to be elucidated to develop effective chemotherapies eliminating blasts and improving clinical outcome.
In summary, our current results provide evidence implicating ATRA as an inhibitor of Nrf2 transcriptional activity in AML cells during ATO challenge that correlates with enhanced cytotoxicity via a mechanism involving RARα activation. Future research will explore the molecular mechanism involving Nrf2 inhibition by ATRA in response to ATO, as well as the role of mutations present in the LBD domain of RARα and/or PML–RARα, to predict resistance during ATO-ATRA therapy.
Change history
26 August 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ablain J, Leiva M, Peres L, Fonsart J, Anthony E, de The H (2013) Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J Exp Med 210: 647–653.
Alarifi S, Ali D, Alkahtani S, Siddiqui MA, Ali BA (2013) Arsenic trioxide-mediated oxidative stress and genotoxicity in human hepatocellular carcinoma cells. Onco Targets Ther 6: 75–84.
Apfel C, Bauer F, Crettaz M, Forni L, Kamber M, Kaufmann F, LeMotte P, Pirson W, Klaus M (1992) A retinoic acid receptor alpha antagonist selectively counteracts retinoic acid effects. Proc Natl Acad Sci USA 89: 7129–7133.
Chen Y, Lin-Shiau S, JK LIN (1998) Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J Cell Physiol 177: 324–333.
Chendamarai E, Balasubramanian P, George B, Viswabandya A, Abraham A, Ahmed R, Alex AA, Ganesan S, Lakshmi KM, Sitaram U, Nair SC, Chandy M, Janet NB, Srivastava VM, Srivastava A, Mathews V (2012) Role of minimal residual disease monitoring in acute promyelocytic leukemia treated with arsenic trioxide in frontline therapy. Blood 119: 3413–3419.
Chou C, Jie C, Kenedy A, Jones RJ, Trush MA, Dang CV (2004) Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc Natl Acad Sci USA 101: 4578–4583.
Davison K, Cote S, Mader S, Miller WH (2003) Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines. Leukemia 17: 931–940.
Duprez E, Benoit G, Flexor M, Lillehaug JR, Lanotte M (2000) A mutated PML/RARA found in the retinoid maturation resistant NB4 subclone, NB4-R2, blocks RARA and wild-type PML/RARA transcriptional activities. Leukemia 14: 255–261.
Goto E, Tomita A, Hayakawa F, Atsumi A, Kiyoi H, Naoe T (2011) Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood 118: 1600–1609.
Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50: 323–354.
Gupta K, Chakrabarti A, Rana S, Ramdeo R, Roth BL, Agarwal ML, Tse W, Agarwal MK, Wald DN (2011) Securinine, a myeloid differentiation agent with therapeutic potential for AML. PLoS One 6: e21203.
Heasman SA, Zaitseva L, Bowles KM, Rushworth SA, Macewan DJ (2011) Protection of acute myeloid leukaemia cells from apoptosis induced by front-line chemotherapeutics is mediated by haem oxygenase-1. Oncotarget 2: 658–668.
Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, Wang ZY (1998) Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72: 567–572.
Iland HJ, Bradstock K, Supple SG, Catalano A, Collins M, Hertzberg M, Browett P, Grigg A, Firkin F, Hugman A, Reynolds J, Di Iulio J, Tiley C, Taylor K, Filshie R, Seldon M, Taper J, Szer J, Moore J, Bashford J, Seymour JF Australasian Leukaemia and Lymphoma Group (2012) All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120: 1570–1580.
Isbister GK, Dawson AH, Whyte IM (2004) Arsenic trioxide poisoning: a description of two acute overdoses. Hum Exp Toxicol 23: 359–364.
Kamimura T, Miyamoto T, Harada M, Akashi K (2011) Advances in therapies for acute promyelocytic leukemia. Cancer Sci 102: 1929–1937.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nuñez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G Nomenclature Committee on Cell Death 2009 (2009) Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ 16: 3–11.
Lang E, Grudic A, Pankiv S, Bruserud O, Simonsen A, Bjerkvig R, Bjørås M, Bøe SO (2012) The arsenic-based cure of acute promyelocytic leukemia promotes cytoplasmic sequestration of PML and PML/RARA through inhibition of PML body recycling. Blood 120: 847–857.
Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD (2013) Arsenic inhibits autophagic flux activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol 33: 2436–2446.
Ma Q (2012) Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426.
Marasca R, Zucchini P, Galimberti S, Leonardi G, Vaccari P, Donelli A, Luppi M, Petrini M, Torelli G (1999) Missense mutations in the PML/RARalpha ligand binding domain in ATRA-resistant As(2)O(3) sensitive relapsed acute promyelocytic leukemia. Haematologica 84: 963–968.
Miller WH Jr., Schipper HM, Lee JS, Singer J, Waxman S (2002) Mechanisms of action of arsenic trioxide. Cancer Res 62: 3893–3903.
Morales AA, Gutman D, Cejas PJ, Lee KP, Boise LH (2009) Reactive oxygen species are not required for an arsenic trioxide-induced antioxidant response or apoptosis. J Biol Chem 284: 12886–12895.
Nakamura S, Nagano S, Nagao H, Ishidou Y, Yokouchi M, Abematsu M, Yamamoto T, Komiya S, Setoguchi T (2013) Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PLoS One 8: e69466.
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin TT, Yu VL, Miller DK (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376: 37–43.
Park JH (2012) ATRA plus arsenic gets another "A" in APL. Blood 120: 1535–1536.
Patatanian E, Thompson DF . Retinoic acid syndrome: a review (2008) J Clin Pharm Ther 33: 331–338.
Rahman I, Kode A, Biswas SK (2006) Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc 1: 3159–3165.
Rushworth SA, MacEwan DJ (2011) The role of Nrf2 and cytoprotection in regulating chemotherapy resistance of human leukemia cells. Cancers 3: 1605–1621.
Rushworth SA, Zaitseva L, Langa S, Bowles KM, MacEwan DJ (2010) FLIP regulation of HO-1 and TNF signalling in human acute myeloid leukemia provides a unique secondary anti-apoptotic mechanism. Oncotarget 1: 359–366.
Saeed S, Logie C, Stunnenberg HG, Martens JH (2011) Genome-wide functions of PML-RARalpha in acute promyelocytic leukaemia. Br J Cancer 104: 554–558.
Sekeres MA, Maciejewski JP, Erba HP, Afable M, Englehaupt R, Sobecks R, Advani A, Seel S, Chan J, Kalaycio ME (2011) A Phase 2 study of combination therapy with arsenic trioxide and gemtuzumab ozogamicin in patients with myelodysplastic syndromes or secondary acute myeloid leukemia. Cancer 117: 1253–1261.
Sharma M, Khan H, Thall PF, Orlowski RZ, Bassett RL Jr, Shah N, Bashir Q, Parmar S, Wang M, Shah JJ, Hosing CM, Popat UR, Giralt SA, Champlin RE, Qazilbash MH (2012) A randomized phase 2 trial of a preparative regimen of bortezomib, high-dose melphalan, arsenic trioxide, and ascorbic acid. Cancer 118: 2507–2515.
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY (1997) Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89: 3354–3360.
Tallman MS, Altman JK (2009) How I treat acute promyelocytic leukemia. Blood 114: 5126–5135.
Valenzuela M, Perez-Perez G, Corvalan AH, Carrasco G, Urra H, Bravo D, Toledo H, Quest AF (2010) Helicobacter pylori-induced loss of the inhibitor-of-apoptosis protein survivin is linked to gastritis and death of human gastric cells. J Infect Dis 202: 1021–1030.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114: 937–951.
Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Li Y, Luo L, Hayes JD, Wang XJ, Tang X (2013) RXR alpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res 73: 3097–3108.
Wang L, Weng CY, Wang YJ, Wu MJ (2011) Lipoic acid ameliorates arsenic trioxide-induced HO-1 expression and oxidative stress in THP-1 monocytes and macrophages. Chem Biol Interact 190: 129–138.
Wang XJ, Hayes JD, Henderson CJ, Wolf CR (2007) Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci USA 104: 19589–19594.
Wetzler M, Andrews C, Ford LA, Tighe S, Barcos M, Sait SN, Block AW, Nowak NJ, Baer MR, Wang ES, Baumann H (2011) Phase 1 study of arsenic trioxide, high-dose cytarabine, and idarubicin to down-regulate constitutive signal transducer and activator of transcription 3 activity in patients aged <60 years with acute myeloid leukemia. Cancer 117: 4861–4868.
Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, Zhou GB, Tong JH, Zhang Y, Wu JH, Hu HY, de Thé H, Chen SJ, Chen Z (2010) Arsenic trioxide controls the fate of the PML-RARalphaoncoprotein by directly binding PML. Science 328: 240–243.
Zhou DC, Kim SH, Ding W, Schultz C, Warrell RP Jr., Gallagher RE (2002) Frequent mutations in the ligand-binding domain of PML-RAR alpha after multiple relapses of acute promyelocytic leukemia: analysis for functional relationship to response to all-trans retinoic acid and histone deacetylase inhibitors in vitro and in vivo. Blood 99: 1356–1363.
Acknowledgements
We are grateful to Dr Mario P Tshan for providing the NB4 and NB4-R2 cell lines. This work was supported by Grant FNRS-Télévie n° 7.4577.11.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Valenzuela, M., Glorieux, C., Stockis, J. et al. Retinoic acid synergizes ATO-mediated cytotoxicity by precluding Nrf2 activity in AML cells. Br J Cancer 111, 874–882 (2014). https://doi.org/10.1038/bjc.2014.380
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.380
Keywords
This article is cited by
-
Athelia rolfsii Exopolysaccharide Protection Against Kidney Injury in Lead-Exposed Mice via Nrf2 Signaling Pathway
Biological Trace Element Research (2023)
-
All-trans retinoic acid enhances the cytotoxic effect of decitabine on myelodysplastic syndromes and acute myeloid leukaemia by activating the RARα-Nrf2 complex
British Journal of Cancer (2023)
-
Hypoxic Hepatocellular Carcinoma Cells Acquire Arsenic Trioxide Resistance by Upregulating HIF-1α Expression
Digestive Diseases and Sciences (2022)
-
All-trans retinoic acid and arsenic trioxide fail to derepress the monocytic differentiation driver Irf8 in acute promyelocytic leukemia cells
Cell Death & Disease (2017)
-
Co-delivery with nano-quercetin enhances doxorubicin-mediated cytotoxicity against MCF-7 cells
Molecular Biology Reports (2016)
