
Abstract
Background:
We conducted a phase I study in patients with advanced solid tumours to identify the recommended dose, assess pharmacokinetics (PK), pharmacodynamic activity and preclinical antitumour efficacy of the combination of sirolimus and gemcitabine.
Methods:
Nineteen patients were treated with sirolimus 2 or 5 mg daily and gemcitabine 800 or 1000 mg m−2 on days 1 and 8. Dose escalation depended on dose-limiting toxicity (DLT) rate during the first 3-week period. Paired skin biopsies were evaluated for phosphorylated S6 (pS6) as marker of mTOR (mammalian target of rapamycin) inhibition. Pharmacokinetics and preclinical evaluation of efficacy using two different sarcoma cell lines and leiomyosarcoma xenografts were also conducted.
Results:
Three DLTs were observed: grade 3 transaminitis, grade 3 thrombocytopenia and grade 4 thrombocytopenia. Common treatment-related adverse events included anaemia, neutropenia, thrombocytopenia and transaminitis. Pharmacodynamic analyses demonstrated mTOR inhibition with sirolimus 5 mg and PK showed no influence of sirolimus concentrations on gemcitabine clearance. In vitro and in vivo studies suggested mTOR pathway hyperactivation by gemcitabine that was reversed by sirolimus. Tumour growth in leiomyosarcoma xenografts was dramatically inhibited by the treatment.
Conclusions:
Recommended dose was sirolimus 5 mg per 24 h plus gemcitabine 800 mg m−2. Antitumour activity in preclinical sarcoma models and mTOR signalling inhibition were observed. A phase II study is currently ongoing.
Similar content being viewed by others


Main
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that plays a central role in the phosphatidyl inositol 3′-kinase (PI3K)-AKT signalling pathway (Aoki et al, 2001; Sabatini, 2006). Activation of mTOR by different environmental and nutritional stimuli triggers transduction of proliferative signals by the phosphorylation of two key downstream effectors, the p70 S6 kinase and the eukaryotic initiation factor 4E binding protein 1 (4EBP-1; Janus et al, 2005). These proteins are involved in the biosynthesis of ribosomes and translation of mRNA necessary for normal cell-cycle regulation (Mamane et al, 2006). The correlation between mTOR pathway abnormalities and carcinogenesis has been extensively reported (Shaw and Cantley, 2006; Hernando et al, 2007). Indeed, up to half of all human tumours have been found to be somehow driven by alterations in the mTOR pathway (Vivanco and Sawyers, 2002; Xu et al, 2004). In addition, it is also critical in some tumour microenvironment processes such as angiogenesis (Viñals et al, 1999; Guba et al, 2002; Hudson et al, 2002; Humar et al, 2002; Mayerhofer et al, 2002; Land and Tee, 2007). Therefore, targeting mTOR is a rational therapeutic approach in human cancer. Sirolimus, also known as rapamycin, was one of the first compounds able to inhibit mTOR (Wiederrecht et al, 1995). It is a macrolide that prevents the phosphorylation of S6 and 4EBP-1 and therefore their activation (Brown et al, 1994; Faivre et al, 2006). Some of its derivatives, namely everolimus, temsirolimus and ridaforolimus, have been successfully assessed in phase III trials in different malignancies (Hudes et al, 2007; Motzer et al, 2008; Yao et al, 2011; Baselga et al, 2012; Demetri et al, 2013).
Gemcitabine is a pyrimidine analogue that targets cells undergoing DNA synthesis and blocks progression of cells from G1 to S-phase (Elnaggar et al, 2012). It is currently used in a vast spectrum of tumours either alone or in combination thanks to its favourable toxicity profile (Gesto et al, 2012).
Combination of sirolimus with gemcitabine has been reported to increase apoptosis in vitro and enhance antitumoural activity in vivo on different epithelial tumours (Grünwald et al, 2002; Mondesire et al, 2004). Specifically in sarcomas, an in vitro study in leiomyosarcoma cell lines has shown that this combination has a synergic effect in extracellular-signal-regulated kinases (ERK 1/2) inhibition, producing a dramatic effect in cell cycle (Merimsky et al, 2007). However, no studies in xenograft sarcoma models have been published to date. Nevertheless, response in a patient affected by leiomyosarcoma has been reported (Merimsky, 2004) suggesting that this combination may have profound effects on these malignancies.
This phase I trial was designed to determine the recommended dose (RD), safety profile, pharmacokinetic (PK) parameters and pharmacodynamic activity of the combination of sirolimus and gemcitabine. Preclinical antitumour efficacy both in vitro and in vivo was also evaluated.
Materials and methods
Patient selection
To be enrolled in this study, patients had to meet the following eligibility criteria: diagnosis of advanced solid tumour that have progressed or are ineligible for standard treatment, no prior treatment with mTOR inhibitors or gemcitabine, Eastern Cooperative Oncology Group performance status (ECOG PS) 0–1, either measurable or evaluable disease and age ⩾18 and ⩽70 years. The upper limit of age was established due to the increased risk of toxicity often seen in some elderly patients. Adequate bone marrow, hepatic and renal function were mandatory and were defined as: absolute neutrophil count ⩾1.5 × 109 l−1, platelets ⩾100 × 109 l−1, bilirubin, aspartate aminotranferase (AST), alanine aminotransferase and creatinine ⩽1.5 × upper limit of normal and creatinine clearance ⩾60 ml min−1. Patients with a history of other previous malignancies diagnosed or treated in the past 5 years (except basal cell skin carcinoma, adenocarcinoma in situ of the uterine cervix and superficial bladder cancer) and known central nervous system metastases were considered ineligible. Other exclusion criteria were treatment with experimental drugs within 30 days prior, pregnancy or lactancy, presence of active infection or any concomitant serious disease.
All patients signed written informed consent and the study was conducted according to local and national ethical review board approval, the Declaration of Helsinki and standards of Good Clinical Practice.
Study design and drug dosage, escalation and administration
Sirolimus was administered as a continuous daily oral dose (2 or 5 mg) starting on day 2 of cycle 1 until progression or intolerance. Gemcitabine was administered intravenously at a fixed-dose rate of 10 mg m−2 min−1 on days 1 and 8 of each cycle. The duration of each cycle was 21 days. A maximum of six cycles of gemcitabine per patient were allowed. Single agent sirolimus was continued after six planned cycles of gemcitabine in the absence of progressive disease (PD) and good tolerance. Protocol was amended according to pharmacodynamic results and a new dose level was added (Table 1).
The trial was performed using a standard 3+3 dose-escalation phase I design with cohorts of 3–6 patients. If less than one-third of patients at a dose level experienced a dose-limiting toxicity (DLT), dose escalation continued. If more than one-third but less than two-thirds of patients at a dose level had a DLT, three additional patients were enrolled at that same dose level. If two-thirds or more of patients at a dose level experienced a DLT, the dose was considered toxic and the next cohort of patients was included at the next lower dose level. Dose escalation within a patient was not permitted. Patients were withdrawn from study treatment when there was evidence of PD, unacceptable toxicity or consent withdrawal.
Routine clinical and laboratory assessments were conducted on a weekly basis. Toxicity was graded using the National Cancer Institute Common Toxicity Criteria version 3.0 (NCI-CTCAE v3.0). Dose-limiting toxicity was defined as any of the following within 3 weeks after the administration of the first cycle: absolute neutrophil count <0.5 × 109 l−1 over ⩾5 days or associated with fever ⩾38.5°C, platelets <50 × 109 l−1, any grade 3–4 non-haematological toxicity (excluding nausea and vomiting non-refractory to antiemetic treatment) or skin rash grade 2 related to treatment and not controlled with support medication. Maximum tolerated dose (MTD) was defined as the highest dose level in which two or more patients experienced DLT. Next lower dose level was considered as RD.
In order to assess tumour response to treatment, thorax–abdomen–pelvis CT scans were performed every 6 weeks and Response Evaluation Criteria in Solid Tumors version 1.0 (RECIST v1.0) were used (Therasse et al, 2000).
Pharmacokinetics
Gemcitabine concentrations were measured at days 1 and 24 of the study and PK sampling was performed at 0.5, 1, 2.5, 4, 8, 10 and 24 h after the start of the infusion, which was ranged from 0.95 to 3.28 h.
Gemcitabine PK blood samples were collected in polypropylene tubes with EDTA, which contained tetrahydrouridine to inactivated gemcitabine degradation. After plasma separation by centrifugation, samples were stored at −80°C until analysis. An Alliance 2695 (Milford, MA, USA) separations module and photodiode array detector, with Empower 2 software (Waters, Milford, MA, USA) to online data acquisition were used. Separation was performed on a Nova Pak C18 cartridge column, (Waters), which was maintained at 30°C. The mobile phases consisted of solutions of 5% (v/v) heptane sulfonic acid and methanol, and were delivered following a flow rate of 1 ml min−1. Gemcitabine and its internal standard (2-desoxicitidina) were extracted from plasma samples by protein precipitation followed liquid–liquid extraction. This HPLC method was validated using quality control samples and standard of calibration obtained from spiked blank plasma samples with different concentrations of gemcitabine. Intra-assay and inter-day imprecision and accuracy was evaluated with the control samples plasma at three concentrations in four days and the values obtained were <10% and 8%, respectively. The limit of quantification (LLOQ) was 200 μg l−1 and measurements were linear from 200 to 20 000 μg l−1 (r2=0.99).
Sirolimus concentrations were measured at day 21 of the study before both gemcitabine and sirolimus dose administrations (pre-dose concentrations). Sirolimus PK blood samples were collected into plasma tubes with EDTA-K3 (Vacuette, Kremsmünster, Austria) and stored at −80°C until analysis. An Acquity UPLC integrated measurement system (Waters) was used. Separation was performed on a MassTraK TDM C18 cartridge column, 2.1 × 10 mm (Waters), which was maintained at 55°C. The mobile phases, consisted of solutions of ammonium acetate 2 mM and 0.1% (v/v) formic acid either in water or in methanol, and were delivered following a flow rate of 0.4 ml min−1. Detection was carried out using an Acquity TQD tandem-quadrupole mass spectrometer equipped with a Z-spray electrospray ionisation source (Waters) operating in positive mode. Sirolimus and its internal standard ([13C2D4]-everolimus) were detected in multiple reaction monitoring mode using mass-to-charge (m/z) transition of 931.9→864.4 and 981.9→914.4, respectively. The MassTrak immunosuppressants XE RUO kit provided by Waters was used. Intra-assay and inter-day coefficients of variation, accuracy and relative measurement errors ranged from 7.8% to 10.0%, 8.9% to 12.4%, −8.7% to −6.0% and −5.0% to 15.0%, respectively. The limit of quantification was 1.7 μg l−1 and the measurement interval was linear between 1.7 and 31.1 μg l−1 (r2=0.996).
The population PK model development and simulations were performed with the nonlinear mixed-effects modelling (NONMEM) software, version 7.2 (ICON Development Solutions, Ellicott City, MD, USA) using the subroutine ADVAN3 TRANS4 (user-defined non-linear model). To statistically distinguish between nested models, the difference in the MOFV8 (minimum objective function value) was used because this difference is approximately χ2 distributed. A significance level of P<0.005 that corresponded to a difference in MOFV of 7.879 for 1 degree of freedom was considered. Additionally, to the diagnostic plots used for evaluation during model building development with Xpose version 4.0 (Division of Pharmacokinetics and Drug Therapy, Uppsala University, Uppsala, Sweden), an internal validation was performed. The bootstrap method with replacement was used to assess the stability of the final model and to construct confidence intervals of PK parameters using the PsN-Toolkit (version 3.2.4; Division of Pharmacokinetics and Drug Therapy, Uppsala University, Uppsala, Sweden).
Pharmacodynamics
Paired skin biopsies were planned for every patient: at baseline and 21 days after first dose administration. In order to assess mTOR pathway inhibition, immunohistochemistry of phosphorylated S6 at Ser235/236 (pS6) #4858 was performed in formalin-fixed paraffin-embedded sections of skin samples using a 1 : 50 dilution of a rabbit polyclonal antibody (from Cell Signaling Technology, Danvers, MA, USA). Then, qualitative changes in the expression of pS6 were assessed.
In vitro study
Two sarcoma cell lines acquired from Cell Lines Service (CLS, Eppelheim, Germany) were used to assess the in vitro efficacy of the treatment: SKLMS-1 and SW982 (leiomyosarcoma and synovial sarcoma, respectively). Both cell lines were cultured in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) and were incubated at 37°C in a humidified atmosphere of 5% CO2 in air.
Cell proliferation assay
Sirolimus and gemcitabine were diluted in cell medium at 20 ng ml−1 and 100 nM, respectively and then cells were treated with both drugs separately, sequentially and in combination for 48 h. Dimethyl sulfoxide (DMSO) was added to cultures as control. Cell proliferation and cell death were determined by the trypan blue exclusion assay.
Western blot
SKLMS-1- and SW982-treated cells were lysed with radioimmunoprecipitation assay buffer containing protease inhibitors (1 mmol l−1 phenylmethylsulfonyl fluoride, 10 mg ml−1 aprotinin, and 10 mg ml−1 leupeptin) and the lysates were centrifuged at 13 000 × g, at 4°C, for 30 min. Lysate aliquots (50 μg) were resolved by 10% SDS–PAGE and transferred onto nitrocellulose membranes. After blocking with 5% skimmed milk in PBS containing 0.2% Tween 20 (Dallas, TX, USA) at room temperature for 1 h, membranes were incubated overnight at 4°C with the appropriate primary antibody (cleaved caspase 3 #9661, native S6 #2217, and pS6 #4858 from Cell Signaling Technology). Blots were then incubated at room temperature for 1 h with a horseradish peroxidase-conjugated secondary antibody and the peroxidase activity was detected by enhanced chemiluminescence (Pierce, Rockford, IL, USA) following the instructions of the manufacturer. Immunodetection of α-tubulin was used as a loading reference.
In vivo study
An in vivo xenograft model was established by subcutaneous injection of 3.5 × 106 SKLMS-1 cells suspended in 100 μl of saline in athymic nude mice (BALB/cnu/nu) from Harlan (Indianapolis, IN, USA). Animal care and procedures were followed according to the Institutional Guidelines for the Care and Use of Laboratory Animals. Once tumours reached 100 mm3, groups of five mice were treated with sirolimus 2.5 mg kg−1 and gemcitabine 60 mg kg−1 followed by sirolimus 2.5 mg kg−1 after 24 h. All treatments were administered in intraperitoneal manner for 2 weeks (sirolimus once daily and gemcitabine once weekly). An additional group of five mice were treated with DMSO as controls. Tumours were measured every 2 days with calipers, and toxicity was monitored by weight loss. Mice were killed once tumours reached 2500 mm3 (or after manifestation of morbidity) and tumours were removed and stored in 4% paraformaldehyde. Immunohistochemistry was performed in formalin-fixed paraffin-embedded sections from tumour samples. Phosphorylated S6 was detected with a 1 : 50 dilution of a rabbit polyclonal antibody #4858 (from Cell Signaling Technology).
Results
Patient characteristics
From June 2010 to September 2011, 19 patients were enrolled in a single centre. All patients were assessable for toxicity and efficacy. Demographics characteristics are shown in Table 2. All patients except one had received prior chemotherapy treatment. Median number of previous lines was 2.5 (range 0–6) and 7 (37%) patients had radiation therapy before enrolment in the study. A total of 77 cycles of the study regimen were administered. Median number of cycles per patient was 4 (range 1–6).
Safety
All 19 patients were evaluable for DLT. Initially, the three dose levels planned were evaluated. One patient experienced DLT consisting in grade 3 transaminitis at dose level 2 and two patients experienced DLT at dose level 3 consisting in grade 3 thrombocytopenia and grade 4 thrombocytopenia, respectively. Thus, MTD was reached at dose level 3. However, the pharmacodynamic analysis performed in the 13 patients treated at those dose levels revelled poor mTOR pathway inhibition at doses <5 mg of sirolimus. Therefore, an amendment was performed including a new dose level under the reached MTD consisting of sirolimus 5 mg and gemcitabine 800 mg m−2 (dose level 2.A). At this dose level, no DLT was observed and it was established as the RD (Table 1).
The majority of side effects reported were grade 1–2. The most commonly observed treatment-related events were haematological: anaemia (84%; n=16), neutropenia (68%; n=13) and thrombocytopenia (68%; n=13). The most frequent non-haematological toxicities were raised AST (58%; n=11), raised GGT (47%; n=9), hypercholesterolaemia (47%; n=9), anorexia (47%; n=9) and mucositis (42%; n=8). In general, toxicity was mild and easily manageable. No pulmonary toxicity was reported. Three patients required dose reduction of sirolimus, being grade 3 thrombocytopenia the reason in two cases and grade 2 fever in one case. Gemcitabine dose reduction was required in two patients due to grade 4 anaemia and grade 2 transaminitis, respectively. Toxicity is summarised in Table 3.
Pharmacokinetics and pharmacodynamics
Since gemcitabine is a drug with well-known activity against a large number of malignancies, we designed the study to determine whether the addition of sirolimus has any influence on its PK. Data from all 19 patients were used in the PK analysis. The effects of gender, age, weight (WGT), body surface area (BSA) and sirolimus through concentrations were assessed on gemcitabine PK at day 21. Demographic characteristics and sirolimus trough concentrations are summarised in Table 4. Correlation between WGT/BSA and height (HGT) was found.
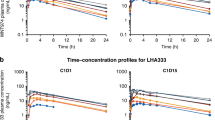
The plasma concentration vs time profiles of gemcitabine at days 1 and 21 are displayed in Figure 1. It should be noted that quantifiable gemcitabine concentrations were found up to 2.5–4 h post administration in both occasions. The PK of gemcitabine after intravenous infusion of 10 mg m−2 min−1 in the target population was best described by a two-open-compartment model with first-order elimination. All recorded covariates were tested in the PK parameters, plasma clearance (CL) and central compartment distribution volume (Vc), with NONMEM, but no statistically significant relationship could be identified in any case. No statistically significant effect of anthropometric covariates (WGT, HGT and BSA) and age on the PK parameters was found (P>0.05) and no specific trends were observed between CL or Vc values and sirolimus concentrations (Supplementary Figure 1). The estimated PK parameters with final model (NONMEN) listed in Supplementary Table 1 were in agreement with those previously reported in the literature (Keith et al, 2003; Lin et al, 2004). Between-patient variability could be associated to CL (14.6%) and Vc (98.2%), meanwhile between-occasion variability could be to Vc (47.1%).

(A ) Observed gemcitabine plasma concentrations ( μ g l−1) vs time (h) after intravenous infusion of 10 mg m−2 min−1 on day 1. (B) Observed gemcitabine plasma concentrations (μg l−1) vs time (h) after intravenous infusion of 10 mg m−2 min−1 on day 21.
Immunohistochemistry of pS6 in patients’ paired skin biopsies showed significant inhibition of mTOR at RD (Supplementary Figure 2). Weaker staining of pS6 was achieved with 5 mg (dose levels 2.A and 3) compared to 2 mg.
Efficacy
Two patients achieved partial response (PR): one patient at dose level 2.A (colon adenocarcinoma) and the other one at dose level 3 (uterine cervix cancer). Nine patients experienced stable disease (SD) as best response that lasted >12 weeks and in three cases, the duration of the stabilisation was at least 6 months.
In vitro study results
Cell proliferation assay results
Both cell lines were sensitive to gemcitabine and sirolimus. Interestingly, higher cell death rate was observed in both cell lines with the sequential treatment administering first gemcitabine and 24 h later sirolimus than with the inverse order or with the administration of both drugs at the same time (data not shown).
Western blot results
We used cleaved caspase 3 as apoptosis marker to assess the in vitro efficacy of the combination. Results showed that the greatest activation of apoptosis was achieved with the sequential treatment administering gemcitabine first followed by sirolimus 24 h later (Figure 2A).

(A ) Western blot cleaved caspase 3. The greatest cleavage of caspase 3 was achieved when treatment was administered in a sequential manner: first gemcitabine followed by sirolimus 24 h later. (B) Western blot pS6 and S6. The activation of S6 observed when cells were treated with gemcitabine alone was reversed with the addition of sirolimus. G=gemcitabine; S=sirolimus; V=control.
We assessed by western blot phosphorylation of S6 as a marker of mTOR activity. Although the non-phosphorylated forms had no relevant changes with the treatment, pS6 was highly induced when cells were treated with gemcitabine alone. This induction was clearly reversed when sirolimus was added (Figure 2B).
In vivo study results
Xenograft model was established using SKLMS-1 cells. According to in vitro results, treatment was administered in a sequential fashion (first gemcitabine and 24 h later sirolimus). Tumour growth was strongly inhibited with the sequential combination of the two drugs compared to Control and to each drug alone (Figure 3).

SKLMS-1 xenograft tumour growth. t-Test: *P⩽0.03; **P⩽0.0001. Leiomyosarcoma xenograft tumour growth was strongly inhibited by the combination treatment. GEM=gemcitabine; SIR=sirolimus.
Immunohistochemistry results
Strong pS6 staining in tumours treated with gemcitabine alone was observed. In contrast, that staining was dramatically absent in tumours treated with the combination, indicating that the addition of sirolimus is able to reverse pS6 induction also in vivo (Figure 4).

Immunohistochemistry of pS6 in leiomyosarcoma xenograft samples. Sirolimus is able to reverse the hyperactivation of the mTOR pathway caused by gemcitabine in leiomyosarcoma xenografts. GEM=gemcitabine; SIR=sirolimus.
Discussion
This study demonstrates that the combination of sirolimus and gemcitabine is feasible and safe, allowing administration of active doses of both agents and achieving mTOR pathway inhibition even in heavily pretreated patients. The most common adverse events registered were haematological, but they were generally mild and easily manageable. Other mild toxicities observed were raised liver enzymes, hypercholesterolaemia, anorexia and mucositis, all of them usually related to either sirolimus or gemcitabine in monotherapy, but modifications in the treatment schedule or dose were not necessary in almost any case. Furthermore, the toxicity profile showed no synergistic effects in these adverse events with the combination of the two drugs. Transaminitis grade 3 and thrombocytopenia grades 3 and 4 where the DLTs found, all of them are relatively common and expected in patients treated with gemcitabine. No unexpected toxicity appeared with the treatment. Moreover, PK showed no effects of sirolimus concentrations on gemcitabine clearance. This favourable profile leads us to recommend dose level 2.A (sirolimus 5 mg per 24 h plus gemcitabine 800 mg m−2) as the optimal dose due to its well-proved safety record.
In addition, the preclinical study also showed encouraging results. Thus, the in vitro study showed that caspase 3 cleavage was more evident when cells were treated sequentially (gemcitabine before sirolimus) than administering both drugs simultaneously. Therefore, a clear pro-apoptotic induction as a result of this combination is responsible for the dramatic effect on tumour survival. Sequential administration of drugs, including sirolimus, as a cancer therapeutic strategy has been used elsewhere (Iacovelli et al, 2013; Rosa et al, 2013). mTOR inhibition results in downregulation of several antiapoptotic proteins such as Bcl-xL and Mcl-1 (Tirado et al, 2005; Faber et al, 2014). Thus, sirolimus addition sequentially after gemcitabine may prevent resistance to this drug through antiapoptotic pathway activation. In agreement with this hypothesis, several reports demonstrate that inhibition of antiapoptotic bcl-2 family members sensitises tumour cells to gemcitabine (Schniewind et al, 2004; Zhang et al, 2011). In contrast, one of the main effects of mTOR inhibition is G1 arrest (Carew et al, 2011) that makes cells less prone to be damaged by gemcitabine. This hypothesis is being currently tested in the laboratory. On the other hand, we found both in vitro and in vivo that S6 was activated when cells were treated with gemcitabine alone but such activation dramatically reversed when sirolimus was added, correlating with the efficacy of the combinatory treatment. These interesting data suggest hyperactivation of mTOR pathway as a cellular mechanism of defence triggered by gemcitabine that can be reversed with the addition of sirolimus. This brand new finding opens an exciting line of investigation worth exploring. Furthermore, xenograft tumour growth was dramatically reduced with the combined treatment and pharmacodynamic analysis showed an effective mTOR inhibition at RD, making this therapeutic strategy even more promising.
Combination of an mTOR inhibitor with conventional chemotherapy with gemcitabine could be a way to improve the efficacy of either of the agents alone in different tumour types such as pancreatic cancer, renal cell cancer or sarcomas. Specifically, in sarcomas, positive results with mTOR inhibitors have been reported. Thus, sirolimus and its derived temsirolimus have shown activity in perivascular epithelioid cell tumours (PEComas), a specific subtype of mesenchymal tumour (Italiano et al, 2010; Wagner et al, 2010). Moreover, it has been recently published in a positive phase III trial in sarcomas with the mTOR inhibitor ridaforolimus. This double-blind, placebo-controlled phase III trial randomised 702 sarcoma patients who had achieved CR, PR, or SD after 1, 2, or 3 lines of chemotherapy to receive placebo or ridaforolimus as maintenance treatment. Ridaforolimus showed signs of activity, inducing a mean 1.3% decrease in target lesion size vs a 10.3% increase with placebo. In addition, it achieved a statistically significant improvement in PFS compared to placebo in both independent and per investigator assessment. However, the magnitude of that improvement was very modest (median PFS 17.7 weeks vs 14.6 weeks per independent review; Demetri et al, 2013). These results, positive but excessively limited, suggest some important conclusions: mTOR inhibitors are active in sarcomas but the best therapeutic strategy is still unknown. Thereafter, combination treatments with mTOR inhibitors and cytotoxic drugs (like the one assessed in this study) are a promising alternative that deserve further investigation.
In conclusion, this phase I trial of the combination of sirolimus and gemcitabine demonstrated that this regimen is feasible and safe. Moreover, it showed signs of activity both in vitro and in vivo. In addition, mTOR inhibition was achieved at RD and PK analysis showed no influence of sirolimus on gemcitabine clearance. Further studies to assess the activity of this combination are warranted and a phase II trial in sarcomas is ongoing (ClinicalTrials.gov identifier NCT01684449).
Change history
26 August 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aoki M, Blazek E, Vogt PK (2001) A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA 98 (1): 136–141.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN (2012) Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366 (6): 520–529.
Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369 (6483): 756–758.
Carew JS, Kelly KR, Nawrocki ST (2011) Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol 6 (1): 17–27.
Demetri GD, Chawla SP, Ray-Coquard I, Le Cesne A, Staddon AP, Milhem MM, Penel N, Riedel RF, Bui-Nguyen B, Cranmer LD, Reichardt P, Bompas E, Alcindor T, Rushing D, Song Y, Lee RM, Ebbinghaus S, Eid JE, Loewy JW, Haluska FG, Dodion PF, Blay JY (2013) Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol 31 (19): 2485–2492.
Elnaggar M, Giovannetti E, Peters GJ (2012) Molecular targets of gemcitabine action: rationale for development of novel drugs and drug combinations. Curr Pharm Des 18 (19): 2811–2829.
Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT, Boisvert JL, Milano RJ, Roper J, Kodack DP, Jain RK, Corcoran RB, Rivera MN, Ramaswamy S, Hung KE, Benes CH, Engelman JA (2014) mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov 4 (1): 42–52.
Faivre S, Kroemer G, Raymond E (2006) Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 5 (8): 671–688.
Gesto DS, Cerqueira NM, Fernandes PA, Ramos MJ (2012) Gemcitabine: a critical nucleoside for cancer therapy. Curr Med Chem 19 (7): 1076–1087.
Grünwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M (2002) Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res 62 (21): 6141–6145.
Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK (2002) Rapamycin inhibits primary and metastatic tumour growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 8 (2): 128–135.
Hernando E, Charytonowicz E, Dudas ME, Menendez S, Matushansky I, Mills J, Socci ND, Behrendt N, Ma L, Maki RG, Pandolfi PP, Cordon-Cardo C (2007) The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med 13 (6): 748–753.
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ, Trial GA (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356 (22): 2271–2281.
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22 (20): 7004–7014.
Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ (2002) Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J 16 (8): 771–780.
Iacovelli R, Cartenì G, Sternberg CN, Milella M, Santoni M, Di Lorenzo G, Ortega C, Sabbatini R, Ricotta R, Messina C, Lorusso V, Atzori F, De Vincenzo F, Sacco C, Boccardo F, Valduga F, Massari F, Baldazzi V, Cinieri S, Mosca A, Ruggeri EM, Berruti A, Cerbone L, Procopio G (2013) Clinical outcomes in patients receiving three lines of targeted therapy for metastatic renal cell carcinoma: results from a large patient cohort. Eur J Cancer 49 (9): 2134–2142.
Italiano A, Delcambre C, Hostein I, Cazeau AL, Marty M, Avril A, Coindre JM, Bui B (2010) Treatment with the mTOR inhibitor temsirolimus in patients with malignant PEComa. Ann Oncol 21 (5): 1135–1137.
Janus A, Robak T, Smolewski P (2005) The mammalian target of the rapamycin (mTOR) kinase pathway: its role in tumourigenesis and targeted antitumour therapy. Cell Mol Biol Lett 10 (3): 479–498.
Keith B, Xu Y, Grem JL (2003) Measurement of the anti-cancer agent gemcitabine in human plasma by high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 785 (1): 65–72.
Land SC, Tee AR (2007) Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem 282 (28): 20534–20543.
Lin NM, Zeng S, Ma SL, Fan Y, Zhong HJ, Fang L (2004) Determination of gemcitabine and its metabolite in human plasma using high-pressure liquid chromatography coupled with a diode array detector. Acta Pharmacol Sin 25 (12): 1584–1589.
Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N (2006) mTOR, translation initiation and cancer. Oncogene 25 (48): 6416–6422.
Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C (2002) BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood 100 (10): 3767–3775.
Merimsky O (2004) Targeting metastatic leiomyosarcoma by rapamycin plus gemcitabine: an intriguing clinical observation. Int J Mol Med 14 (5): 931–935.
Merimsky O, Gorzalczany Y, Sagi-Eisenberg R (2007) Molecular impacts of rapamycin-based drug combinations: combining rapamycin with gemcitabine or imatinib mesylate (Gleevec) in a human leiomyosarcoma model. Int J Oncol 31 (1): 225–232.
Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, Meric-Bernstam F (2004) Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res 10 (20): 7031–7042.
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A, Group R-S (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372 (9637): 449–456.
Rosa R, Damiano V, Nappi L, Formisano L, Massari F, Scarpa A, Martignoni G, Bianco R, Tortora G (2013) Angiogenic and signalling proteins correlate with sensitivity to sequential treatment in renal cell cancer. Br J Cancer 109 (3): 686–693.
Sabatini DM (2006) mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 6 (9): 729–734.
Schniewind B, Christgen M, Kurdow R, Haye S, Kremer B, Kalthoff H, Ungefroren H (2004) Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int J Cancer 109 (2): 182–188.
Shaw RJ, Cantley LC (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441 (7092): 424–430.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216.
Tirado OM, Mateo-Lozano S, Notario V (2005) Rapamycin induces apoptosis of JN-DSRCT-1 cells by increasing the Bax: Bcl-xL ratio through concurrent mechanisms dependent and independent of its mTOR inhibitory activity. Oncogene 24 (20): 3348–3357.
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2 (7): 489–501.
Viñals F, Chambard JC, Pouysségur J (1999) p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem 274 (38): 26776–26782.
Wagner AJ, Malinowska-Kolodziej I, Morgan JA, Qin W, Fletcher CD, Vena N, Ligon AH, Antonescu CR, Ramaiya NH, Demetri GD, Kwiatkowski DJ, Maki RG (2010) Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835–840.
Wiederrecht GJ, Sabers CJ, Brunn GJ, Martin MM, Dumont FJ, Abraham RT (1995) Mechanism of action of rapamycin: new insights into the regulation of G1-phase progression in eukaryotic cells. Prog Cell Cycle Res 1: 53–71.
Xu G, Zhang W, Bertram P, Zheng XF, McLeod H (2004) Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR pathway in common human tumors. Int J Oncol 24 (4): 893–900.
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Öberg K RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group (2011) Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 364 (6): 514–523.
Zhang C, Cai TY, Zhu H, Yang LQ, Jiang H, Dong XW, Hu YZ, Lin NM, He QJ, Yang B (2011) Synergistic antitumor activity of gemcitabine and ABT-737 in vitro and in vivo through disrupting the interaction of USP9X and Mcl-1. Mol Cancer Ther 10 (7): 1264–1275.
Acknowledgements
We thank Laura Lagares-Tena for her help with the preclinical studies and Pedro Alía and Ana Clopés for their help with the PK analyses. This work was supported by Grants TRA-163 from Spanish Ministry of Health (to XG) and PI12/01908 from Instituto de Salud Carlos III (to XG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Martin-Liberal, J., Gil-Martín, M., Sáinz-Jaspeado, M. et al. Phase I study and preclinical efficacy evaluation of the mTOR inhibitor sirolimus plus gemcitabine in patients with advanced solid tumours. Br J Cancer 111, 858–865 (2014). https://doi.org/10.1038/bjc.2014.370
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.370
Keywords
This article is cited by
-
In vitro drug sensitivity (IDS) of patient-derived primary osteosarcoma cells as an early predictor of the clinical outcomes of osteosarcoma patients
Cancer Chemotherapy and Pharmacology (2020)
-
Phase II Study of Gemcitabine Plus Sirolimus in Previously Treated Patients with Advanced Soft-Tissue Sarcoma: a Spanish Group for Research on Sarcomas (GEIS) Study
Targeted Oncology (2018)
-
Bone Sarcomas in Pediatrics: Progress in Our Understanding of Tumor Biology and Implications for Therapy
Pediatric Drugs (2015)
