
Abstract
Background:
Predictive biomarker development is a key challenge for novel cancer therapeutics. We explored the feasibility of next-generation sequencing (NGS) to validate exploratory genomic biomarkers that impact phase I trial selection.
Methods:
We prospectively enrolled 158 patients with advanced solid tumours referred for phase I clinical trials at the Royal Marsden Hospital (October 2012 to March 2013). After fresh and/or archived tumour tissue were obtained, 93 patients remained candidates for phase I trials. Results from tumour sequencing on the Illumina MiSeq were cross-validated in 27 out of 93 patients on the Ion Torrent Personal Genome Machine (IT-PGM) blinded to results. MiSeq validation with Sequenom MassARRAY OncoCarta 1.0 (Sequenom Inc., San Diego, CA, USA) was performed in a separate cohort.
Results:
We found 97% concordance of mutation calls by MiSeq and IT-PGM at a variant allele frequency ⩾13% and ⩾500 × depth coverage, and 91% concordance between MiSeq and Sequenom. Common ‘actionable’ mutations involved deoxyribonucleic acid (DNA) repair (51%), RAS-RAF-MEK (35%), Wnt (26%), and PI3K-AKT-mTOR (24%) signalling. Out of 53, 29 (55%) patients participating in phase I trials were recommended based on identified actionable mutations.
Conclusions:
Targeted high-coverage NGS panels are a highly feasible single-centre technology well-suited to cross-platform validation, enrichment of trials with molecularly defined populations and hypothesis testing early in drug development.
Similar content being viewed by others


Main
The development of biomarkers in modern anticancer drug development is a critically important challenge. Recent successes in targeted therapy have defined specific gene and/or protein pathways dysregulated and ‘driving’ tumourigenic processes (Romond et al, 2005; Fong et al, 2009; Flaherty et al, 2010; Kwak et al, 2010), but lack of molecular selection has also led to drug attrition (Mateo et al, 2013). Of the increasing number of novel cancer therapeutics developed (Workman et al, 2013), few reach successful late-stage trials (Williams et al, 2012), and biomarker selection may be a necessary tool to improve drug development success (Garraway, 2013). We have previously described the importance of biomarker selection for early-phase trials in the pharmacological audit trail (PhAT), both through utilisation of known predictive biomarkers and through hypothesis testing of putative ‘enrichment’ biomarkers (Yap et al, 2010; Garcia et al, 2011).
Next-generation sequencing (NGS) has brought unprecedented opportunities for biomarker development by vastly expanding the capability and feasibility of molecularly characterising tumours (Ross and Cronin, 2011; Tran et al, 2013). However, despite the potential of whole-genome, whole-exome, or whole-transcriptome sequencing to personalise therapeutic selection, these approaches are not yet practical for routine use in early-phase trials. One recent study showed a median time to NGS results of 91 days (range 43–243) (Weiss et al, 2013), which remains an unreasonable wait time for the patient with treatment-refractory cancer. Major challenges also hindering the routine application of NGS include bioinformatic interpretation of enormous quantities of data, incomplete functional understanding of multiple pathway interactions or uncommon mutations, and the need to validate sequenced mutations (Braggio et al, 2013).
Nevertheless, phase I trial candidates and physicians highly support biomarker selection (Miller et al, 2013), hoping that rational trial selection will improve the odds of response and relevance to cancer treatment. Challenges to testing of biomarkers in the phase I population include a highly heterogeneous population with a mixture of tumour types, heavy pre-treatment burden, multiple sites of disease likely to be molecularly heterogeneous, and varied life expectancy (Carden et al, 2010). The promise of NGS-guided therapeutic selection will depend on overcoming these challenges in a reasonable timeframe with relevance to therapeutic selection of trials (Arkenau et al, 2008).
We sought to pilot the use of targeted, high-coverage NGS technologies at the Royal Marsden Hospital Drug Development Unit (RMH-DDU) and The Institute of Cancer Research (ICR) by testing NGS panels with targeted regions of interest (ROIs) directly relevant to our portfolio of phase I trials. The key issue of validation of genomic mutations was explored through cross-platform comparison of results obtained from two NGS technologies as well as a mass spectrometry-based method. Secondary issues of archived vs fresh tissue sampling (Carter et al, 2012; Spencer et al, 2013), tumour heterogeneity (Gerlinger et al, 2012), and the feasibility of real-time application of this approach in the phase I clinic were explored, including translating NGS results into potentially relevant clinical trials.
Materials and methods
Patient consent and enrolment
Institutional research ethics board approval was obtained. Patients referred for experimental therapy October 2012 to March 2013 at the RMH-DDU were considered for study enrolment. Eligibility criteria were patients with metastatic or locally advanced cancer, age ⩾18 years, referral for early-phase clinical trials, fitness for clinical trials (Eastern Cooperative Group Performance Status (ECOG-PS) 0–2, no major comorbidity requiring active management, no grade 3–4 laboratory abnormalities), and signed informed consent for genetic analysis of tumours. Patient demographic and disease data were collected and entered into an anonymised database with linking accession numbers.
Tissue collection and processing
Tissue analyses were conducted at The ICR Cancer Biomarkers Laboratory according to Human Tissue Act requirements. Archived tumour material (tissue blocks or slides) from primary and/or metastatic samples were retrieved from across 58 referral centres in the UK. If deemed technically safe, fresh tumour tissue biopsies were performed with patient consent by an experienced interventional radiologist. Sample identification including site, collection time, and referring institution were recorded. Fresh tissue samples were paraffin embedded and processed further in the same manner as archived tissue. Tissue samples were reviewed by the histopathology team after haematoxylin and eosin (H&E) staining. The region of tumour was then marked by a pathologist for coring. All samples were labelled with anonymised tissue identification.
DNA extraction and quality control
The DNA was manually extracted using the QIAamp DNA FFPE Tissue kit (Qiagen, Venlo, Limburg, Netherlands) following the manufacturer’s protocol. Eluted DNA was measured using nanodrop and Quant-iT high-sensitivity Picogreen double-stranded DNA (dsDNA) Assay Kit (Invitrogen, ThermoFisher Scientific Corp., Waltham, MA, USA), according to the manufacturer’s recommendations. DNA-QC was performed using the Illumina FFPE QC kit (WG-321-1001; see Supplementary Methods).
Illumina MiSeq sequencing
The Illumina MiSeq TruSeq Amplicon Cancer Panel (TSACP) is a highly multiplexed NGS assay covering 212 ROIs in 48 cancer-related genes (Supplementary Table 1), in which 2631 mutations are represented at least twice in the Catalogue of Somatic Mutations in Cancer (COSMIC). Targeted sequencing was performed following the manufacturer’s protocol on samples that passed QC parameters (see Supplementary Methods). Bioinformatic analyses were performed utilising the MiSeq Reporter Software MCS 2.2.0, RTA 1·17·28·0 and Nextgene (from Biogene, Kimbolton, Cambs, UK).
Ion Torrent Personal Genome Machine (PGM) sequencing
The Ion AmpliSeq Cancer Hotspot Panel v2 contains a single pool of primers used to perform multiplexed PCR for preparation of amplicon libraries covering 207 ROIs in 50 cancer-related genes (Supplementary Tables 1 and 2, Figure 1A and B), in which 2175 mutations are represented at least twice in COSMIC. Twenty-seven randomly selected samples from the prospective cohort were selected to compare MiSeq and PGM sequencing. Targeted sequencing was performed following the manufacturer’s protocol (see Supplementary Methods). Bioinformatic analyses were performed using the PGM Torrent Suite software (version 3·4·2) including a Torrent Variant Caller for single-nucleotide polymorphisms and indel variants across a reference BED file, and Nextgene (from Biogene) for comparison with the MiSeq data.

Patient and sample flow diagram. Abbreviations: IT-PGM=Ion Torrent Personal Genome Machine; NGS=next-generation sequencing.
Sequenom OncoCarta v1.0 sequencing
The OncoCarta v1.0 panel comprises 24 multiplex assays that detect 238 mutations in 19 oncogenes (Supplementary Table 1). Twenty-five archived tumour samples from patients referred between 2009 and 2012 who were previously enrolled into a molecular characterisation study were analysed by the Sequenom OncoCarta v1.0 panel (Sequenom Inc.) and MiSeq TSACP (Illumina, San Diego, CA, USA). The samples were processed as described in the methods above for DNA extraction, quantification, QC and sequencing on the MiSeq TSACP. Sequenom OncoCarta v1.0 sequencing was performed as per manufacturer’s instructions (see Supplementary Methods).
Mutation report for multidisciplinary tumour board
DNA mutations reported by each sequencing platform were entered into an anonymised database that was linked to patient and sample data. To minimise false-positive reporting, somatic mutations were only reported if a minimum of 500 × coverage depth had been achieved with a variant allele frequency of at least 5%. Mutations were also coded with results of functional knowledge from the COSMIC database and prior literature review. Mutation data from multiple tumour samples from a single patient were linked. Patient mutation reports were generated containing identifying data, tumour primary, histopathology summary, type of sample(s) (FFPE, fresh, or other), sequencing platform(s) used, mutation(s) detected (gene, frequency, and prior description in literature), and potential functional significance of mutations. ‘Actionability’ of mutations was defined by a literature search of each gene covered by the MiSeq and PGM panels, with potential therapeutics having entered late-stage preclinical or clinical stage (Supplementary Table 3).
Phase I clinical trial allocation
Patient cases were discussed at multidisciplinary rounds including oncologists, radiologists, histopathology staff, nurses, and biomedical scientists, integrating information from the patient mutation report. Only mutations previously reported in COSMIC were considered actionable, and relevance of phase I clinical trials to the identified mutations was discussed. Specific clinical trial information was presented to patients provided that they fit eligibility criteria for participation, and while it was disclosed if relevant mutation data influenced the decision for trial allocation, the experimental nature of this approach was emphasised. Patients ineligible for mutation-relevant clinical trials were offered phase I trials without required selection biomarkers if available, or transferred back to the care of the referring health care team.
Results
Characteristics of patients analysed by NGS
Overall, 207 patients were seen in consultation at the RMH-DDU for phase I trials between October 2012 and March 2013, of which 158 patients (76%) initially met study inclusion (Figure 1). Tumour tissue was collected in 118 patients (75%); 103 archived samples alone, 9 with both fresh tissue (including ascites or pleural fluid) and archival tissue collected, and 7 with a fresh tissue biopsy without archived sample. Median age of tumour samples was 3 years. Upon obtaining tumour tissue, 22 patients were excluded as they were no longer eligible or interested in a phase I clinical trials, and another 3 patients had tissue samples without sufficient tumour for analysis.
In total, 93 patients remained eligible for phase I trials and were processed by NGS (Table 1). The most common tumour primary types were ovarian/peritoneal (n=22, 24%), colorectal (n=19, 20%), breast (n=7, 8%) and bladder (n=6, 76%); 92% of patients had metastatic disease, most commonly to the lungs (45%) and lymph nodes (41%). Two patients had previously treated stable brain metastases; 98% had ECOG-PS 0–1, and 80% had a favourable Royal Marsden Hospital Prognostic Index (RMH-PI; Olmos et al, 2012). Patients had received a median of 2 prior lines of therapy and 33% had been exposed to 3 or more lines of treatment.
Mutations detected by Illumina MiSeq TSACP
Out of 93 patients, 85 (91%) had samples that passed QC for sequencing. Sample QC parameters were not affected adversely by tumour blockage or referral centre (n=58) (Figure 2), although the best QC scores were obtained from fresh tissue sampled in 2013 at site 32 (RMH-DDU). Three hundred and ninety-six mutations were detected from 69 of the 85 samples (81%) that passed QC, while 16 (19%) patients had no mutations detected despite excellent tumour QC parameters (Supplementary Table 4). In total, 232 of these 396 (59%) mutations had been previously described as oncogenic in the COSMIC database; 69 out of 396 (17%) mutations were unreported but impacting an amino acid previously described to be mutated in cancer; 95 out of 396 (24%) were mutations not previously described. Most frequently mutated genes were TP53 (22%), PIK3CA (12%), APC (11%), KRAS (9%), ATM (6%), and FBXW7 (5%) (Figure 3), although a substantial proportion of the mutations seen in ATM and FBXW7 had not been previously described as oncogenic. Mutations in BRAF (3%), MET (3%), KIT (3%), EGFR (3%), ERBB2 (2%), and AKT1 (1%) were also detected.

Effect of blockage and site of block retrieval on quality control parameters for MiSeq. (A) Distribution of DNA-QC scores among samples run on the MiSeq. (B) The effect of blockage on QC scores, showing the best QC scores for fresh tissue obtained in 2013. (C) A plot of QC scores by referring hospital shows that sufficient quality tumour DNA can be extracted from nearly all submitted tumour blocks despite varying laboratory practices. Abbreviations: DNA=deoxyribonucleic acid; QC=quality control.

Histogram of mutations detected across samples by Illumina MiSeq TSACP (prospective cohort). In total, 396 mutations were detected in 70 out of 85 (82%) patients that passed DNA-QC parameters. The majority of mutations were previously described in COSMIC (232 out of 396, 59%), while a substantial proportion of mutations was not previously described (95 out of 396, 24%) or only described as a similar mutation in the same amino acid in COSMIC (69 out of 396, 17%). Abbreviations: AA=amino acid; COSMIC=Catalogue of Somatic Mutations in Cancer; DNA-QC=Deoxyribonucleic acid quality control; TSACP=TruSeq Amplicon Cancer Panel.
Mutations were detected at a frequency similar to the expected mutation rates in the ovarian and colorectal cancer cohorts (Supplementary Table 5), considering the limited sample size. Histological subtypes influenced mutation rates, for example, in the ovarian cancer cohort where high rates of KRAS and PIK3CA mutations were detected: in the low-grade serous carcinoma group (seven patients, 38%), 3 out of 7 (43%) had a KRAS mutation and 4 out of 7 (57%) had PIK3CA mutation, and in both a clear cell and a mucinous ovarian carcinoma, concurrent KRAS and PIK3CA mutations were detected.
Blinded analysis of mutations from patients with multiple samples shows the ability of MiSeq TSACP to detect mutations reproducibly between primary tumour, synchronous metastases, and metachronous metastases (Table 2; patients 1–5 and 7). Interestingly the detected variant allele frequencies were strikingly similar between multiple tumour samples from the same patient. Only in one patient mutations were not reproducibly detected between samples (patient 6), in which all mutations were found at low variant allele frequencies (5–10%).
Cross-validation of MiSeq results by IT-PGM ACP
Concordance of IT-PGM results with MiSeq results was very high for overlapping ROIs, with 21 out of 27 patient samples (78%) demonstrating 100% concordance of mutation results, and only 2 out of 27 (7%) patient samples having fully discordant results (Table 3). Overall, 84% of mutations detected by MiSeq TSACP were also detected by PGM AmpliSeq at the pre-defined coverage depth (500 × ) and frequency parameters (5%). Notably, 14 out of 15 (93%) of discordant mutation calls had allele frequencies of 12% or less, whereas 38 out of 39 (97%) of concordant mutation calls had allele frequencies of 13% or greater. Variant allele frequencies of gene mutations detected on both NGS platforms were strikingly similar.
Cross-validation of MiSeq results with Sequenom OncoCarta v1.0
Concordance of Sequenom OncoCarta v1.0 mutations with MiSeq results was very high for overlapping mutation ‘hotspots’, with 19 out of 21 (91%) of patient samples demonstrating 100% concordance of results (Supplementary Table 6). Overall, 14 out of 14 (100%) of mutations detected by both platforms had variant allele frequencies >13% on MiSeq. Discordant mutation calls were an EGFR S725I mutation (patient 8) and ABL1 G250E mutation (patient 18) detected by Sequenom but not by MiSeq, and a BRAF V600E mutation (patient 20) detected by MiSeq but not by Sequenom.
Translating actionable mutations into phase I trial selection
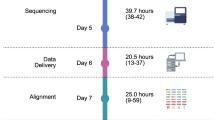
Actionable mutations were detected in 100% of colorectal, breast, and lung cancer samples, 86% of ovarian cancer samples, and 74% of the overall patient population (Table 4 and Supplementary Table 7). The most commonly implicated therapeutic targets were in DNA repair (51%), RAS-RAF-MEK (35%), Wnt (26%), and PI3K-AKT-mTOR (24%) signalling. The median time from receipt of tissue in the laboratory to the reporting of sequencing results was 5 days. Patient tumour mutation reports were generated in 93 out of 93 (100%) patients sequenced and all were presented at the multidisciplinary tumour board for decisions regarding patient allocation to clinical trials.
Overall, 53 out of 93 (57%) patients were allocated to 58 clinical trials between October 2012 and June 2013; 29 out of 53 patients (55%) who participated in clinical trials had trials suggested based on a relevant mechanism of action for the identified ‘actionable’ mutation (Table 5), whereas 29 out of 63 (46%) patients with identified actionable mutations participated in matched trials. Conversely, lack of availability of an open clinical trial slot at the time of tumour board discussion was the prime reason (50%) why patients with actionable mutations detected could not participate in a matched study. Of patients ‘matched’ to trial, 14 (48%) targeted PI3K-AKT-mTOR, 6 (20%) targeted RAS-RAF-MEK, 5 (18%) targeted DNA repair, and 4 (14%) targeted insulin-like growth factor (IGF) or AGC kinases.
Although objective response data will be analysed once a larger experience with matched treatment is gained and the relevant trials are reported, examples of responses to matched treatment include: (1) a patient with an oestrogen receptor positive, Her2 amplification-negative breast cancer, found to have a highly prevalent ERBB2 V777L mutation known to activate Her2 signalling. Treatment with a dual mTORC1/mTORC2 inhibitor led to a Response Evaluation Criteria in Solid Tumours (RECIST) partial response for 8 months (Bose et al, 2013). (2) A patient with high-grade serous ovarian carcinoma of the ovarian with two activating mutations in PIK3CA (P104L, W552G), both rare mutations in the p85 domain and the other in the catalytic domain, each reported only twice previously. This patient was treated with a dual mTOR-PI3K oral inhibitor and achieved a RECIST partial response but discontinued therapy after 5 months due to drug-related toxicity.
Discussion
In our study, 158 patients were consented to study, 118 (75%) had tissue retrieved/collected, 93 (59%) were eligible for phase I trials and had NGS done, and 85 (54%) had tumour DNA that passed quality control for NGS. Of these eligible and sequenced 85 patients, 15 (18%) had no mutations detected, 7 (10%) had no ‘actionable’ mutation, and 63 (74%) had ‘actionable’ mutations detected, of which 29 out of 63 (46%) participated in a matched clinical trial. Therefore, the overall rate of success in matching patients to clinical trial within our trial portfolio was 29 out of 158 (18%) considering all of the feasibility hurdles. This single-centre pilot study demonstrates that targeted, high-coverage NGS can deliver biomarker-enriched patient populations for early-phase trials, allowing for early hypothesis testing as part of PhAT. Rapid cross-validation of detected mutations was feasible by different NGS technologies with very high concordance using paraffin tissue. Overall, 74% of successfully sequenced patients had ‘actionable’ mutations, of which 46% of patients with actionable mutations were ‘matched’ to trial.
A major advantage to targeted sequencing on the MiSeq or IT-PGM platforms is that ROIs can be customised to match the specific clinical trial portfolio of the drug development unit, allowing a focussed evaluation of potential biomarkers of interest. We also found that the advantages included scalability according to patient volumes, moderate material costs (∼$250 per patient sample run for MiSeq TSACP, excluding tissue processing and labour; see Supplementary Table 2), and a fast turnaround time with a median tissue receipt to reporting of results of 5 days. Previous targeted sequencing in the phase I setting have reported on a low proportion of patients participating in trials (Tran et al, 2013), and while broader genomic approaches to sequencing have been advocated as a means of personalising treatment (Roychowdhury et al, 2011), these are currently too resource and time intensive for the majority of treatment-refractory patients considering phase I trials. Whole-genome sequencing analyses frequently require months of bioinformatics analyses before the finalised reports are available. Moreover, these approaches may be better utilised to define the biological underpinnings of tumours that have an exceptional response to therapy in view of the current costs of whole-genome sequencing (Kwak et al, 2010).
The reliance of NGS on software for data analyses to interpret the large amount of information generated makes it critically important to validate detected mutations (Ross and Cronin, 2011). Cross-validation of mutations by different NGS sequencing panels was possible due to significant overlap in ROIs. Prior technical studies have suggested variant frequency cut-offs of 10% and minimum of 250 × depth coverage (Singh et al, 2013); we chose 5% variant frequency and a minimum of 500 × coverage depth for reporting, which is conservative given the manufacturer’s labelling of 2% sensitivity. On cross-platform analysis we found discrepancies occurred almost exclusively at low variant allele frequencies (<13%), suggesting that in real-life clinical samples, validation with highly sensitive, multiplexed and unbiased methods such as NGS are feasible and appropriate (Hadd et al, 2013).
In order to conduct this study, we received tumour samples from hospitals across the UK. Therefore a significant limitation to the quality of the DNA analysed included the prior handling of tumour tissue. The quality of the DNA extracted from an archival sample is highly dependent on how the sample has been handled before, during and after fixation and embedding. Great care should be taken during the fixation process to prevent overfixation (not >24 h) that would result in more extensive crosslinking and make extraction of nucleic acids of good quality more difficult. During the embedding process it is important that the specimen is fully dehydrated to prevent degradation and stored appropriately. Storage at 4 °C compared to room temperature has been shown to better preserve nucleic acids.
Our study shows that there is significant variability in the quality control parameters based on the site of archived tumour retrieval. Nevertheless, the majority of archived tumour samples still achieved good quality control and could be successfully sequenced. Archived FFPE tissue was the main tissue source because of availability and patient preference. The formalin fixation process does not appear to alter NGS mutation calls if DNA quality is adequate (Spencer et al, 2013), and in this study identical mutations were detected among multiple patient samples (primary and metastatic).
On-study fresh tumour biopsies are ideal specimens for molecular characterisation, but the challenges are well documented in the literature; these include informed patient consent, appropriate selection of biopsy sites, logistical coordination of radiological guided biopsies, monitoring of patients post tumour biopsies, adequate staffing for real-time processing and analysis of tumour specimens, and adequate analytical validation of biomarker assays. The safety of biopsies are generally excellent, and at our site we performed superficial, lymph node or liver biopsies on-site but referred thoracic biopsies to a hospital with thoracic surgical support.
The use of liquid biopsy is showing promise through detection of circulating tumour DNA (ctDNA), but not all types of cancer release equal amounts (Bettegowda et al, 2014). Tumour heterogeneity and evolution are important factors to consider in the treatment-refractory patient (Collisson et al, 2012; Gerlinger et al, 2012; Swanton, 2012), and less invasive tissue sources such as ctDNA and malignant fluid (as explored in this study), may be necessary tools in patients unable or unwilling to undergo a biopsy (Agulnik et al, 2006; Perkins et al, 2012; Murtaza et al, 2013).
Conclusion
Overall, we show that targeted, multiplexed NGS panels assessing customised ROIs can quickly and cost-effectively allow drug development units to enrich their trials with patients harbouring mutations of interest as suggested by the PhAT. Challenges remain in the clinical implementation of NGS technologies, but in this study a high proportion of patients had actionable mutation results (63 out of 85; 74%) and nearly half of the patients with actionable mutations were directed to relevant trial options (29 out of 63, 46%). Future directions will include further customisation of ROIs to match our trial portfolio, a greater emphasis on prospective biopsy sampling including ctDNA, and application of biomarker data from exceptional responders to inform trial allocation.
Change history
26 August 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agulnik M, Oza AM, Pond GR, Siu LL (2006) Impact and perceptions of mandatory tumor biopsies for correlative studies in clinical trials of novel anticancer agents. J Clin Oncol 24 (30): 4801–4807.
Arkenau HT, Graeven U, Kubicka S, Grothey A, Englisch-Fritz C, Kretzschmar A, Greil R, Freier W, Seufferlein T, Hinke A, Schmoll HJ, Schmiegel W, Porschen R AIO Colorectal Study Group (2008) Oxaliplatin in combination with 5-fluorouracil/leucovorin or capecitabine in elderly patients with metastatic colorectal cancer. Clin Colorectal Cancer 7 (1): 60–64.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Diaz LA Jr (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6 (224): 224ra224.
Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Ellis MJ (2013) Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 3 (2): 224–237.
Braggio E, Egan JB, Fonseca R, Stewart AK (2013) Lessons from next-generation sequencing analysis in hematological malignancies. Blood Cancer J 3: e127.
Carden CP, Sarker D, Postel-Vinay S, Yap TA, Attard G, Banerji U, de Bono JS (2010) Can molecular biomarker-based patient selection in phase I trials accelerate anticancer drug development? Drug Discov Today 15 (3-4): 88–97.
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Getz G (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30 (5): 413–421.
Collisson EA, Cho RJ, Gray JW (2012) What are we learning from the cancer genome? Nat Rev Clin Oncol 9 (11): 621–630.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363 (9): 809–819.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361 (2): 123–134.
Garcia VM, Cassier PA, de Bono J (2011) Parallel anticancer drug development and molecular stratification to qualify predictive biomarkers: dealing with obstacles hindering progress. Cancer Discov 1 (3): 207–212.
Garraway LA (2013) Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol 31 (15): 1806–1814.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366 (10): 883–892.
Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, Latham GJ (2013) Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn 15 (2): 234–247.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Iafrate AJ (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363 (18): 1693–1703.
Mateo J, Ong M, Tan DS, Gonzalez MA, de Bono JS (2013) Appraising iniparib, the PARP inhibitor that never was-what must we learn? Nat Rev Clin Oncol 10 (12): 688–696.
Miller FA, Hayeems RZ, Bytautas JP, Bedard PL, Ernst S, Hirte H, Siu LL (2013) Testing personalized medicine: patient and physician expectations of next-generation genomic sequencing in late-stage cancer care. Eur J Hum Genet 22 (3): 391–395.
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Rosenfeld N (2013) Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497 (7447): 108–112.
Olmos D, A’Hern RP, Marsoni S, Morales R, Gomez-Roca C, Verweij J, Kaye SB (2012) Patient selection for oncology phase I trials: a multi-institutional study of prognostic factors. J Clin Oncol 30 (9): 996–1004.
Perkins G, Yap TA, Pope L, Cassidy AM, Dukes JP, Riisnaes R, de Bono JS (2012) Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS One 7 (11): e47020.
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, Wolmark N (2005) Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 353 (16): 1673–1684.
Ross JS, Cronin M (2011) Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol 136 (4): 527–539.
Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Chinnaiyan AM (2011) Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med 3 (111): 111ra121.
Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, Luthra R (2013) Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn 15 (5): 607–622.
Spencer DH, Sehn JK, Abel HJ, Watson MA, Pfeifer JD, Duncavage EJ (2013) Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J Mol Diagn 15 (5): 623–633.
Swanton C (2012) Intratumor heterogeneity: evolution through space and time. Cancer Res 72 (19): 4875–4882.
Tran B, Brown AM, Bedard PL, Winquist E, Goss GD, Hotte SJ, Dancey JE (2013) Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int J Cancer 132 (7): 1547–1555.
Weiss GJ, Liang WS, Demeure MJ, Kiefer JA, Hostetter G, Izatt T, Craig DW (2013) A pilot study using next-generation sequencing in advanced cancers: feasibility and challenges. PLoS One 8 (10): e76438.
Williams RJ, Walker I, Takle AK (2012) Collaborative approaches to anticancer drug discovery and development: a Cancer Research UK perspective. Drug Discov Today 17 (5-6): 185–187.
Workman P, Al-Lazikani B, Clarke PA (2013) Genome-based cancer therapeutics: targets, kinase drug resistance and future strategies for precision oncology. Curr Opin Pharmacol 13 (4): 486–496.
Yap TA, Sandhu SK, Workman P, de Bono JS (2010) Envisioning the future of early anticancer drug development. Nat Rev Cancer 10 (7): 514–523.
Acknowledgements
The authors would like to thank Professor Stan Kaye, Dr Udai Banerji and Dr Rhoda Molife for recruiting the patients to this study, and to Ms Kirsty Moran and Ms Helen Touloui for their invaluable efforts in organisational and logistical support. This work was supported by funding from Cancer Research UK and the Department of Health in the UK through an Experimental Medicine Cancer Centre grant as well as a Biomedical Research Centre grant to the Royal Marsden NHS Foundation Trust.
Author contributions
MO was involved in study conception and design, literature search, ethics submission, patient consent and data collection, data analysis, and manuscript writing. SC was involved in study conception and design, literature search, MiSeq and Ion Torrent sequencing and analysis, data analysis, and manuscript writing. JG contributed to study design, literature search, MiSeq and Ion Torrent sequencing and analysis, data analysis, and manuscript writing. JM was involved in study design, ethics submission, patient consent and data collection, data analysis, and manuscript writing. IF contributed to histopathological analysis. DNR contributed to histopathological analysis. GP contributed to study design. GS contributed to bioinformatics analysis and data storage. TAY was involved in ethics submission, patient consent, data collection, study conception and design. GA was involved in study conception and design. JSdB was involved in study conception and design, ethics submission, patient consent and data collection, data analysis, data interpretation, and manuscript writing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Ong, M., Carreira, S., Goodall, J. et al. Validation and utilisation of high-coverage next-generation sequencing to deliver the pharmacological audit trail. Br J Cancer 111, 828–836 (2014). https://doi.org/10.1038/bjc.2014.350
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.350
Keywords
This article is cited by
-
The AURORA pilot study for molecular screening of patients with advanced breast cancer–a study of the breast international group
npj Breast Cancer (2017)
-
Comparative genetic analysis of a rare synchronous collision tumor composed of malignant pleural mesothelioma and primary pulmonary adenocarcinoma
Diagnostic Pathology (2016)
-
Neueste technologische Entwicklungen für die Analyse von zirkulierender Tumor-DNA
Medizinische Genetik (2016)
