
Abstract
Background:
Current data suggest that platinum-based combination therapy is the standard first-line treatment for biliary tract cancer. EGFR inhibition has proven beneficial across a number of gastrointestinal malignancies; and has shown specific advantages among KRAS wild-type genetic subtypes of colon cancer. We report the combination of panitumumab with gemcitabine (GEM) and oxaliplatin (OX) as first-line therapy for KRAS wild-type biliary tract cancer.
Methods:
Patients with histologically confirmed, previously untreated, unresectable or metastatic KRAS wild-type biliary tract or gallbladder adenocarcinoma with ECOG performance status 0–2 were treated with panitumumab 6 mg kg−1, GEM 1000 mg m−2 (10 mg m−2 min−1) and OX 85 mg m−2 on days 1 and 15 of each 28-day cycle. The primary objective was to determine the objective response rate by RECIST criteria v.1.1. Secondary objectives were to evaluate toxicity, progression-free survival (PFS), and overall survival.
Results:
Thirty-one patients received at least one cycle of treatment across three institutions, 28 had measurable disease. Response rate was 45% and disease control rate was 90%. Median PFS was 10.6 months (95% CI 5–24 months) and median overall survival 20.3 months (95% CI 9–25 months). The most common grade 3/4 adverse events were anaemia 26%, leukopenia 23%, fatigue 23%, neuropathy 16% and rash 10%.
Conclusions:
The combination of gemcitabine, oxaliplatin and panitumumab in KRAS wild type metastatic biliary tract cancer showed encouraging efficacy, additional efforts of genetic stratification and targeted therapy is warranted in biliary tract cancer.
Similar content being viewed by others
Main
Biliary tract cancer (BTC) encompasses both intra- and extrahepatic cholangiocarcinomas and gallbladder carcinoma. The annual incidence is >12 000 cases (Siegel et al, 2013) in the United States with a rising number of intrahepatic cholangiocarcinomas (Patel, 2001). Curative therapy for BTC is surgical resection, however, over 80% of patients present with metastatic disease, for whom treatment options are palliative. Based on the ABC-02 trial, the standard of care for advanced BTC is cisplatin and gemcitabine, which demonstrated significant improvement in median overall survival (OS) of 11.7 months compared to 8.3 months with gemcitabine alone (Valle et al, 2010). Additional trials have evaluated gemcitabine in combination with oxaliplatin with response rates in excess of 30% and favourable toxicity profile, providing another option for first-line therapy (Andre et al, 2004, Pracht et al, 2012).
The epidermal growth factor receptor (EGFR) is broadly expressed in BTC and in vitro studies have suggested the potential efficacy of inhibitors of this pathway (Harder et al, 2009). It has been further suggested that tumour genetics may influence the impact of EGFR inhibition. Preclinical data has demonstrated KRAS wild-type (KRAS WT) human cholangiocarcinoma cell lines show increased sensitivity to cetuximab when compared to KRAS mutant cell lines (Xu et al, 2010). In another study vandetanib, a partial EGFR inhibitor, suppressed growth of TKKK (KRAS WT) BTC cell line, while HuCCT (KRAS mutant) required 10 times the amount of drug for similar effect (Yoshikawa et al, 2009). KRAS mutations are found in 3–50% of tumours depending on tumour location and methodology used to evaluate for mutations in different studies (Suto et al, 2000; Rashid et al, 2002; Ong et al, 2012). The most recent report suggests that KRAS mutations are most common in extrahepatic tumours and that rates for intrahepatic cancers are on the lower end of this spectrum at 9–22% (Voss et al, 2013). Early phase trials evaluating EGFR inhibition in BTC demonstrated clinical activity with observed responses (Philip et al, 2006; Lubner et al, 2010). These early trials, however, were not guided by tumour genetics and the pattern of responses was variable with no clear anatomic or pathologic corollaries identified. Given this, and the paradigm of sensitivity to EGFR blocking antibodies as established in colon cancer (Douillard et al, 2010), we undertook a molecularly driven approach evaluating panitumumab, a fully human EGFR blocking monoclonal, in combination with gemcitabine and oxaliplatin (GEMOX) in KRAS WT BTC.
Materials and Methods
This phase II clinical trial was reviewed and approved by the institutional review boards at the Wilmot Cancer Center, University of Rochester (Rochester, NY, USA) and the Dana-Farber/Harvard Cancer Center (Boston, MA, USA).
All patients provided written informed consent before participation in accordance with institutional and federal guidelines.
At the University of Rochester tumour-derived DNA (isolated from FFPE biopsy material) was assessed for KRAS mutations in codons 12 and 13 via PCR using Roche Light Cycler. At Massachusetts General Hospital Cancer Center, total nucleic acids were extracted from FFPE diagnostic tumour tissue and mutational profiling was performed using a custom modified ABI PRISM SNaPshot Multiplex System on an ABI PRISM 3730 DNA-Analyzer (Life Technologies/Applied Biosystems, Grand Island, NY, USA), as previously described (Dias-Santagata et al, 2010).
Eligibility criteria
Eligibility included histological confirmation metastatic or unresectable KRAS WT biliary tract adenocarcinoma (bile ducts, hepatic duct, cystic duct, common bile duct, ampulla of Vater or gallbladder adenocarcinoma) with measurable disease defined as at least one lesion >2 cm that can be accurately measured with standard imaging techniques. Participants were 18 years of age or older with no history of chemotherapy or anti-EGFR therapy for biliary tract or gallbladder cancer. Eligible patients required an ECOG performance status of 0 or 1, adequate hepatic, renal and haematologic function (serum bilirubin <2.5 mg dl−1, aspartate aminotransferase and alanine aminotransferase <2.5 × institutional upper limit of normal, serum creatinine below upper institutional limits or creatinine clearance >60 ml min−1 per 1.73 m−2, absolute neutrophil count (ANC) >1500 mcl−1, platelet count >100 000 mcl−1 and a life expectancy >3 months. Patients with concurrent malignancies were excluded unless they were treated with curative intent with no known active disease for 3 years prior to enrolment and these include adequately treated non-melanomatous skin cancer or lentigo maligna, treated cervical carcinoma in situ without evidence of disease, prostatic intraepithelial neoplasia without evidence of disease or DCIS without evidence of invasive breast cancer. Patients with known brain metastasis were also excluded. Patients with pre-existing grade 2 or higher peripheral neuropathy were excluded. Prior chemoembolization or radiation to the liver was allowed as long as there was measurable disease outside the radiation area and at least 4 weeks had lapsed since therapy. Women of childbearing potential and men were required to agree to the use adequate contraception and pregnant women were excluded.
Study design and treatment
The trial was designed as an open-label, single-arm phase II study. Eligible patients were treated first with panitumumab at 6 mg kg−1 over 1 h, followed by gemcitabine at 1000 mg m−2 as dose rate infusion at 10 mg m−2 min−1, and then with oxaliplatin at 85 mg m−2 over 2 h, on days 1 and 15 of every 28-day cycle. Patients were screened with computed tomography (CT) scans of the chest, abdomen and pelvis, physical examination, blood chemistries and KRAS evaluation. During treatment patients were assessed prior to therapy on days 1 and 15 of each cycle; CT scans and CA19-9 levels were performed every 8 weeks.
Toxicity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Treatment was discontinued in the event of disease progression, performance status of ⩾3, or participant withdrawal. Panitumumab was held for symptomatic skin or nail-related toxicity or any clinically related ⩾ grade 3 toxicity. When panitumumab was withheld due to skin or nail toxicity the administration of GEMOX was left to the clinical discretion of the treating physician. Gemcitabine and oxaliplatin were held for ANC <1000 mcl−1 or platelet count <75 000 mcl−1, or other ⩾ grade 3 non-haematologic toxicities. Treatment could be delayed for up to 3 weeks to allow for recovery from toxicity, if the patient did not meet re-treatment criteria after a 3-week delay, then the patient will be removed from the study. Antibiotics and steroids were permitted for panitumumab-related rash at the discretion of the treating physician.
The trial was registered at Clinical Trials.gov with the identifier NCT01308840.
Statistical analysis
The primary endpoint was the radiographic response rate by RECIST criteria to GEMOX+panitumumab and secondary endpoints included progression-free survival (PFS), OS and toxicity. A sample size of 30 patients was selected to achieve a power of 80% to detect an absolute difference in response rate of 20% (50% vs 30%) using a one-sided binomial test with a type 1 error set at 0.10. Adverse events and response data were presented as frequencies and percentages. Associated two-sided 95% confidence intervals (CIs) were constructed. Progression-free survival was defined as the time from study enrolment to date of cancer progression or death, whichever occurred first, and OS was defined as the time of enrolment in the study until the date of death from any cause. The distributions of PFS and OS were estimated using the method of Kaplan–Meier. The analysis was performed using intent-to-treat (ITT) principles. All analyses were conducted using SAS software (version 9.3; SAS Institute, Cary, NC, USA).
Results
Patient characteristics
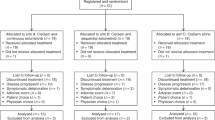
Between 2010 and 2012, 38 patients were enrolled in the trial; of these, 28 were evaluable for efficacy and 31 for toxicity (Figures 1 and 2). Baseline characteristics are shown in Table 1. Three patients were excluded due to having a KRAS mutation and two due to having insufficient tissue for KRAS testing. Of note, a number of patients were selected based on known KRAS status established by clinical broad-based institutional tumour genotyping efforts independent of this trial. Thirty-three patients were allocated to receive treatment but 2 patients did not receive treatment as they were removed from the study due to rapidly progressing symptoms while awaiting KRAS testing. Of the 31 patients who received at least one dose of study drug, 28 patients were evaluable for response because 3 patients did not undergo initial restaging CT evaluation. Reasons for not receiving a second CT evaluation included one patient whose performance status declined rapidly, one patient who was hospitalised during the first cycle followed by an extended treatment delay and then requested to withdraw from the study, and a third patient who developed grade 3 anaemia, hyponatremia, and hyperbilirubinemia and was not able to tolerate additional chemotherapy. Of the included 31 patients, 17 were male and 14 female, the median age was 61 (36–74), 29 patients with metastatic disease and, 2 with locally advanced disease. Baseline characteristics are shown in (Table 1).

Study flow diagram depicts all patients assessed for eligibility and enrolled in the study.

Waterfall plot of percentage of tumour change from baseline in 28 evaluable patients.
Dose intensity
The median number of completed treatment cycles was 6 (range 2–24). The most common indication for dose reductions of gemcitabine was thrombocytopenia and occurred in 32% of patients. Oxaliplatin was dose reduced in 42% of patients secondary to neuropathy. In the 11 patients who completed 10 or more cycles of chemotherapy, 7 patients had to permanently discontinue oxaliplatin for the remainder of their treatment. Patients with preserved performance status of ⩽2 were allowed to remain on gemcitabine and panitumumab after discontinuation of oxaliplatin as long as they maintained clinical benefit (stable disease or objective response). Dose adjustments for panitumumab were infrequent, it was held for one patient who had a grade 3 finger fissures and dose reduced in another patient due to rash.
Efficacy
Of the 31 patients who received treatment, 14 patients had a confirmed partial response resulting in an objective response rate of 45% (95% CI 27–64) in the ITT population (Table 2). Of the 28 patients who had a post treatment follow-up scan, the response rate was 50%. Median OS was 20.3 months (95% CI 9–25 months; Figure 3) and median PFS was 10.6 months (95% CI 5–24 months; Figure 4) in the ITT population.

Kaplan–Meier curve of overall survival (OS).

Kaplan–Meier curve of progression-free survival (PFS).
Toxicity
Thirty-one patients received at least one treatment cycle and were eligible for toxicity assessment. In general, the combination of GEMOX and panitumumab was tolerated reasonably well. Table 3 highlights grade 3 and 4 toxicities. Grade 3 fatigue, anaemia, neuropathy, and electrolyte changes were fairly common; there were no grade 3 or 4 cases of motor toxicity. Patients were not prophylactically treated for EGFR-related rash.
Correlative analysis
EGFR-related rash was seen in 20 patients and hypomagnesemia in 21 patients, there was no statistical correlation between the presence of rash/hypomagnesemia and response.
Discussion
This open-label phase II trial was designed to evaluate the efficacy of the addition of the EGFR inhibitor panitumumab to standard systemic therapy among selected patients with a KRAS WT allele. The chemotherapeutic backbone of GEMOX was chosen given the favourable side-effect profile, convenient schedule (once every 2 weeks) and similar efficacy when compared to gemcitabine and cisplatin (Harder et al, 2006; Verderame et al, 2006; Andre et al, 2008). We found a response rate of 50%, in evaluable patients and 45% in patients who received at least one dose of study drug. The median PFS was 10.6 months and median OS 20.3 months. Furthermore, the combination of gemcitabine, oxaliplatin and panitumumab was well tolerated with manageable grade 3 and 4 toxicities.
There are three randomised trials evaluating targeted EGFR inhibition in BTC and these have offered limited understanding on the impact of KRAS in response to EGFR therapy. BTC is a heterogeneous group of tumours with varying genetic drivers based on geographic region, which may contribute to variable responses to systemic therapy. Such disease heterogeneity makes comparison of trials between geographic regions and with differing composition of diseases (gallbladder, intra- and extrahepatic tumours) challenging. A phase III trial evaluating anti-EGFR therapy among unselected populations assessed the combination of GEMOX with either erlotinib or placebo, the response rate with erlotinib was 30% compared to 16% in the placebo arm, but failed to show a significant increase in PFS or OS (Lee et al, 2012). Only 6 out of 60 patients with evaluable tissue were found to harbour a KRAS mutation limiting any conclusions about the impact of mutations on clinical efficacy. The largest randomised II trial with an EGFR monocloncal antibody (BINGO) enrolled 150 patients to cetuximab and GEMOX vs GEMOX alone, the addition of cetuximab failed to show a statistically significant increase in PFS (Malka et al, 2012). The response rate in the cetuximab–GEMOX arm was 23%, compared to 29% in the GEMOX arm with a trend towards increased OS (12.4 vs 11 months) in the GEMOX arm. Although not statistically significant, there was a trend towards increased PFS in the cetuximab arm, 6 vs 5.3 months. Tumour samples were available for genetic analysis in 50% of patients, KRAS mutations were found in 19% of patients tested, and there was no prognostic or predictive impact based on genetic characterisation identified among this cohort. A third more recent randomised phase II experience with GEMOX and cetuximab found a trend towards increased progression-free and OS in KRAS mutant patients who received cetuximab compared to those who received GEMOX (Chen et al, 2013). This is a surprising finding given the understanding of the molecular biology of mutant RAS signalling as well as paradigms in other tumour types where RAS mutations are predictive of EGFR insensitivity.
In the other phase II marker-driven trial of panitumumab in BTC the response rate was 33% and median PFS 8.3 months (Jensen et al, 2012). In comparison to that trial rates of toxicity with GEMOX and panitumumab were similar; however, we found a lower incidence of rash possibly due to the fact that we did not include capecitabine. The most recent trial evaluating panitumumab in BTC used the chemotherapeutic backbone of gemcitabine and irinotecan resulting in a response rate of 31% with median PFS 9.7 months and OS 12.9 months (Sohal et al, 2013). This study did not exclude patients who were KRAS mutant, and among 17 patients with adequate samples, 7 harboured KRAS mutations.
A summary of published EGFR trials in BTC is outlined in Table 4 (Gruenberger et al, 2010; Rubovszky et al, 2013).
The PFS of 10.6 months and mOS of 20.3 months were the longest among anti-EGFR trials reported to date. The response rate in the intrahepatic cholangiocarcinoma subset (25 pts) was 48%, among the non-intrahepatic cholangiocarcinoma patients there were two responses, one patient with extrahepatic cholangiocarcinoma, and one patient with gallbladder carcinoma. A notable difference in our trial compared to other GEMOX anti-EGFR combinations is that we allowed patients to remain on study receiving gemcitabine and panitumumab even after discontinuation of oxaliplatin secondary to neurotoxicity. Oxaliplatin was discontinued in 25% of patients, who then continued to receive gemcitabine and panitumumab, and it is possible that such an approach, utilising the two agents with little accumulated toxicity as maintenance therapy enabled a relatively long PFS. Given the single-arm design, it is unknown whether the favourable OS outcomes were contributed by including patients with only the KRAS WT signature.
The recent uncovering of the genetic landscape of BTC demonstrates this to be a heterogeneous disease influenced by both anatomic location within the biliary system and on geographic region (Hezel et al, 2010). This will not only make clinical study increasingly challenging, but also much more likely successful, as we should aim to parse out subgroups of patients from each other to test the impact of the new and promising therapeutic targets that have emerged in BTC.
Predicting response to treatment stratified by genetic subtype may spare patients unlikely to respond to targeted therapy unnecessary toxicity, and it may result in cost saving for the healthcare system. At this time the role of anti-EGFR therapy in BTC is modest, with variable results and negative, though relatively small, unselected randomised trials. The prognostic or therapeutic significance of KRAS testing in BTC is unknown highlighting the importance of continued analysis with subsequent trial design. We realise that limitations of our study include single-arm phase II design, which may lead to selection bias and presently survival data is not yet mature. The single group nature of our phase II design precludes definitive conclusions on the added efficacy of panitumumab when added to GEMOX. Nonetheless, here we report the combination of GEMOX and panitumumab with a response rate of 45% in evaluable patients was well tolerated and led to a relatively favourable mPFS and OS. Although further randomised trials excluding KRAS mutant patients may demonstrate a benefit, this would need to be balanced against using efforts and resources in evaluating other new promising targeted agents in BTC and ongoing efforts at genetic stratification to better guide therapeutic approaches.
Change history
29 July 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andre T, Reyes-Vidal JM, Fartoux L, Ross P, Leslie M, Rosmorduc O, Clemens MR, Louvet C, Perez N, Mehmud F, Scheithauer W (2008) Gemcitabine and oxaliplatin in advanced biliary tract carcinoma: a phase II study. Br J Cancer 99: 862–867.
Andre T, Tournigand C, Rosmorduc O, Provent S, Maindrault-Goebel F, Avenin D, Selle F, Paye F, Hannoun L, Houry S, Gayet B, Lotz JP, de Gramont A, Louvet C (2004) Gemcitabine combined with oxaliplatin (GEMOX) in advanced biliary tract adenocarcinoma: a GERCOR study. Ann Oncol 15: 1339–1343.
Chen LCJ, Chao Y, Tsai C, Shan Y, Hsu C, Huang S, Tsou H, Lee K, Chiu C, Rau K, Ho C, Yu M (2013) KRAS mutation status-stratified randomized phase II trial of GEMOX with and without cetuximab in advanced biliary tract cancer (ABTC): The TCOG T1210 trial. 2013 ASCO Annual Meeting J Clin Oncol (suppl; abstr 4018).
Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, Stubbs H, Mcdermott U, Settleman J, Kwak EL, Clark JW, Isakoff SJ, Sequist LV, Engelman JA, Lynch TJ, Haber DA, Louis DN, Ellisen LW, Borger DR, Iafrate AJ (2010) Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med 2: 146–158.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, Blasinska-Morawiec M, Smakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28: 4697–4705.
Gruenberger B, Schueller J, Heubrandtner U, Wrba F, Tamandl D, Kaczirek K, Roka R, Freimann-Pircher S, Gruenberger T (2010) Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: a phase 2 study. Lancet Oncol 11: 1142–1148.
Harder J, Riecken B, Kummer O, Lohrmann C, Otto F, Usadel H, Geissler M, Opitz O, Henss H (2006) Outpatient chemotherapy with gemcitabine and oxaliplatin in patients with biliary tract cancer. Br J Cancer 95: 848–852.
Harder J, Waiz O, Otto F, Geissler M, Olschewski M, Weinhold B, Blum HE, Schmitt-Graeff A, Opitz OG (2009) EGFR and HER2 expression in advanced biliary tract cancer. World J Gastroenterol 15: 4511–4517.
Hezel AF, Deshpande V, Zhu AX (2010) Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol 28: 3531–3540.
Jensen LH, Lindebjerg J, Ploen J, Hansen TF, Jakobsen A (2012) Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Ann Oncol 23: 2341–2346.
Lee J, Park SH, Chang HM, Kim JS, Choi HJ, Lee MA, Jang JS, Jeung HC, Kang JH, Lee HW, Shin DB, Kang HJ, Sun JM, Park JO, Park YS, Kang WK, Lim HY (2012) Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 13: 181–188.
Lubner SJ, Mahoney MR, Kolesar JL, Loconte NK, Kim GP, Pitot HC, Philip PA, Picus J, Yong WP, Horvath L, van Hazel G, Erlichman CE, Holen KD (2010) Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: a phase II Consortium study. J Clin Oncol 28: 3491–3497.
Malka D, Fartoux L, Rousseau V, Trarbach T, Boucher E, de la Fouchardiere C, Faivre SJ, Viret F, Blanc J-F, Assenat E, Hammel P, Louvet C, von Wichert G, Ducreux M, Rosmorduc O, Pignon J-P, Greten TF (2012) Gemcitabine and oxaliplatin (GEMOX) alone or in combination with cetuximab as first-line treatment for advanced biliary cancer: Final analysis of a randomized phase II trial (BINGO). ASCO Meeting Abstracts J Clin Oncol (suppl; abstr 4032).
Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, Mcpherson JR, Allen GE, Ng CC, Wong BH, Myint SS, Rajasegaran V, Heng HL, Gan A, Zang ZJ, Wu Y, Wu J, Lee MH, Huang D, Ong P, Chan-on W, Cao Y, Qian CN, Lim KH, Ooi A, Dykema K, Furge K, Kukongviriyapan V, Sripa B, Wongkham C, Yongvanit P, Futreal PA, Bhudhisawasdi V, Rozen S, Tan P, Teh BT (2012) Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet 44: 690–693.
Patel T (2001) Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 33: 1353–1357.
Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, Donehower RC, Fitch T, Picus J, Erlichman C (2006) Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol 24: 3069–3074.
Pracht M, Le Roux G, Sulpice L, Mesbah H, Manfredi S, Audrain O, Boudjema K, Raoul JL, Boucher E (2012) Chemotherapy for inoperable advanced or metastatic cholangiocarcinoma: retrospective analysis of 78 cases in a single center over four years. Chemotherapy 58: 134–141.
Rashid A, Ueki T, Gao YT, Houlihan PS, Wallace C, Wang BS, Shen MC, Deng J, Hsing AW (2002) K-ras mutation, p53 overexpression, and microsatellite instability in biliary tract cancers: a population-based study in China. Clin Cancer Res 8: 3156–3163.
Rubovszky G1, Láng I, Ganofszky E, Horváth Z, Juhos E, Nagy T, Szabó E, Szentirmay Z, Budai B, Hitre E (2013) Cetuximab, gemcitabine and capecitabine in patients with inoperable biliary tract cancer: a phase 2 study. Eur J Cancer 49: 3806–3812.
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63: 11–30.
Sohal DP, Mykulowycz K, Uehara T, Teitelbaum UR, Damjanov N, Giantonio BJ, Carberry M, Wissel P, Jacobs-Small M, O’dwyer PJ, Sepulveda A, Sun W (2013) A phase II trial of gemcitabine, irinotecan and panitumumab in advanced cholangiocarcinoma. Ann Oncol 24: 3061–3065.
Suto T, Habano W, Sugai T, Uesugi N, Funato O, Kanno S, Saito K, Nakamura S (2000) Aberrations of the K-ras, p53, and APC genes in extrahepatic bile duct cancer. J Surg Oncol 73: 158–163.
Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J Investigators ABCT (2010) Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 362: 1273–1281.
Verderame F, Russo A, di Leo R, Badalamenti G, Santangelo D, Cicero G, Valerio MR, Gulotta G, Tomasello G, Gebbia N, Fulfaro F (2006) Gemcitabine and oxaliplatin combination chemotherapy in advanced biliary tract cancers. Ann Oncol 17 (Suppl 7): vii68–vii72.
Voss JS, Holtegaard LM, Kerr SE, Fritcher EG, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR (2013) Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol 44: 1216–1222.
Xu L, Hausmann M, Dietmaier W, Kellermeier S, Pesch T, Stieber-Gunckel M, Lippert E, Klebl F, Rogler G (2010) Expression of growth factor receptors and targeting of EGFR in cholangiocarcinoma cell lines. BMC Cancer 10: 302.
Yoshikawa D, Ojima H, Kokubu A, Ochiya T, Kasai S, Hirohashi S, Shibata T (2009) Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br J Cancer 100: 1257–1266.
Acknowledgements
This work was funded by a research grant to AFH and AXZ (as the primary investigators) from Amgen, Inc., Thousand Oaks, CA, USA.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
AFH has served as a consultant to Bayer and Novartis and has received speaking honoraria from Celgene, Bayer, and Novartis. MSN has served as a consultant for Bayer. DRB is a consultant for Bio-Reference Laboratories. CF has served as a consultant for Amgen, Takeda, Metamark, Genetics, Genentech, Acceleron and Momenta Pharmaceuticals. AXZ has served as a consultant for Acceleron Pharmaceuticals, Sanofi-aventis, Exelixis, Daiichi Sankyo, Eisai, Celgene and Jennerex. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Hezel, A., Noel, M., Allen, J. et al. Phase II study of gemcitabine, oxaliplatin in combination with panitumumab in KRAS wild-type unresectable or metastatic biliary tract and gallbladder cancer. Br J Cancer 111, 430–436 (2014). https://doi.org/10.1038/bjc.2014.343
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.343
Keywords
This article is cited by
-
Overview of current targeted therapy in gallbladder cancer
Signal Transduction and Targeted Therapy (2020)
-
Tumor-infiltrating mast cells predict prognosis and gemcitabine-based adjuvant chemotherapeutic benefit in biliary tract cancer patients
BMC Cancer (2018)
-
Incidence, risk and prognostic role of anti-epidermal growth factor receptor-induced skin rash in biliary cancer: a meta-analysis
International Journal of Clinical Oncology (2018)
-
Aktuelle Therapieoptionen beim Cholangiokarzinom
Der Gastroenterologe (2017)
