
Abstract
Background:
c-MYC copy number gain (c-MYC gain) has been associated with aggressive behaviour in several cancers. However, the role of c-MYC gain has not yet been determined in lung adenocarcinomas classified by genetic alterations in epidermal growth factor receptor (EGFR), KRAS, and anaplastic lymphoma kinase (ALK) genes. We investigated the clinicopathologic and prognostic significance of c-MYC gain for disease-free survival (DFS) and overall survival (OS) according to EGFR, KRAS, and ALK gene status and stages in lung adenocarcinomas.
Methods:
In 255 adenocarcinomas resected in Seoul National University Bundang Hospital from 2003 to 2009, fluorescence in situ hybridisation (FISH) with c-MYC probe and centromeric enumeration probe 8 (CEP8) was analysed using tissue microarray containing single representative core per each case. EGFR (codon 18 to 21) and KRAS (codon 12, 13, and 61) mutations were analysed by polymerase chain reaction and direct sequencing method from formalin-fixed, paraffin-embedded tissue sections. ALK rearrangement was determined by FISH method. c-MYC gain was defined as >2 copies per nucleus, chromosome 8 gain as ⩾3 copies per nucleus, and gain of c-MYC:CEP8 ratio (hereafter, c-MYC amplification) as ⩾2.
Results:
We observed c-MYC gain in 20% (51 out of 255), chromosome 8 gain in 5.5% (14 out of 255), c-MYC amplification in 2.4% (6 out of 255), EGFR mutation in 49.4% (118 out of 239), KRAS mutation in 5.7% (7 out of 123), and ALK rearrangement in 4.9% (10 out of 205) of lung adenocarcinomas. c-MYC gain was observed in 19% (22 out of 118) of patients with lung adenocarcinomas with an EGFR mutation, but not in any patients with a KRAS mutation, or an ALK rearrangement. c-MYC gain (but not chromosome 8 gain or c-MYC amplification) was an independent poor-prognostic factor in the full cohort of lung adenocarcinoma (P=0.022, hazard ratio (HR)=1.71, 95% confidence interval (CI), 1.08–2.69 for DFS; P=0.032, HR=2.04, 95% CI, 1.06–3.91 for OS), as well as in stage I subgroup (P=0.023, HR=4.70, 95% CI, 1.24–17.78 for DFS; P=0.031, HR=4.65, 95% CI, 1.15–18.81 for OS), and in EGFR-mutant subgroup (P=0.022; HR=2.14; 95% CI, 1.11–4.10 for DFS).
Conclusions:
c-MYC gain (but not chromosome 8 gain or c-MYC amplification) was an independent poor-prognostic factor for DFS and OS in lung adenocarcinomas, both in full cohort and stage I cancer, and possibly for DFS in EGFR-mutant adenocarcinomas. Additional studies are required to determine if patients with lung adenocarcinoma with c-MYC gain are candidates for additional first-line treatment to mitigate their increased risk for disease progression and death.
Similar content being viewed by others


Main
Lung cancer remains the leading cause of cancer-related death worldwide (Jemal et al, 2010; Pao and Girard 2011; Kim et al, 2013b), despite therapeutic advances (Pao and Girard, 2011). About 85% of lung cancer is non-small cell lung cancer (NSCLC), and about 50% is adenocarcinoma (Kim et al, 2013b). Recently, new treatment strategy targeting ‘driver mutations’ including epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) has opened an era of personalised medicine in lung adenocarcinomas (Mok, 2011; Pao and Girard, 2011; Kim et al, 2013b). However, the current standard strategy for the management of lung adenocarcinoma is still early detection and curative surgical resection (Iwakawa et al, 2011). Even with curative surgical resection in early stage, a considerable number of patients eventually recur, and their survival remains unsatisfactory (Sawabata et al, 2010; Shimizu et al, 2013). Therefore, it has been an important issue to identify and validate the molecular prognostic factors affecting recurrence and survival in lung adenocarcinoma patients, especially in those with early stage disease. In this context, various clinicopathologic factors have been investigated (Kudo et al, 2012; Arrieta Rodriguez et al, 2013; Brueckl et al, 2013; Chen et al, 2013; Kawase et al, 2013; Kwon et al, 2013; Nentwich et al, 2013; Sun et al, 2013; Kim et al, 2013a). Although the key genetic alterations have been largely revealed in lung adenocarcinoma (Imielinski et al, 2012), the useful molecular prognostic factors contributing to or accelerating the carcinogenic process, disease progression, or recurrence have not yet been fully understood, especially in stage I disease (Woo et al, 2012; Shimizu et al, 2013).
c-MYC gene is an important member of MYC proto-oncogene containing N-MYC, c-MYC, and L-MYC (Zhang et al, 2010). The c-MYC gene is located at chromosome 8q24, and c-MYC protein functions as a transcription factor regulating cell growth, proliferation, differentiation, and apoptosis (Zhang et al, 2010; Perez et al, 2011; Li et al, 2012). The overexpression of c-MYC protein promotes tumorigenesis by enhancing DNA double-strand breaks, genetic instability, and cell migration, as well as preventing escape from cell cycle (as referenced by Hermeking, 2003; Darcy et al, 2009; Li et al, 2012; Lin et al, 2012). c-MYC protein forms a heterodimer with MAX. The MYC/MAX heterodimer binds to E-box sequences near the promoter region of genes, and enhances the transcription of a wide range of genes (Hermeking, 2003; Lin et al, 2012).
c-MYC protein is elevated in tumours via several ways including translocation and amplification. In Burkitt lymphoma, the tumorigenesis is mainly mediated by t(8;14) translocation involving c-MYC and IGH@ genes, leading to the extremely enhanced proliferating capacity with very short-doubling time in B-lymphoid cells (Swerdllow et al, 2008). Gene amplification or copy number gain of c-MYC have also been documented in non-lymphoid solid tumours including cancers from breast, ovary, prostate, bone, and brain (Liao and Dickson, 2000; Ghadimi et al, 2003; Morrison et al, 2005; Darcy et al, 2009; Perez et al, 2011; Zitterbart et al, 2011; Fromont et al, 2013). In these solid tumours, c-MYC amplification was associated with lymph node metastasis, recurrence, and disease progression to a variable degree (Ghadimi et al, 2003; Darcy et al, 2009; Perez et al, 2011; Fromont et al, 2013). In lung cancer, some early studies revealed frequent c-MYC amplification in small cell lung cancer cell lines (Little et al, 1983; Johnson et al, 1987), and several subsequent studies showed c-MYC amplification or c-MYC copy number gain (c-MYC gain) in NSCLC in animal model or human tumour tissues by using various methods (Kubokura et al, 2001; Rapp et al, 2009; Job et al, 2010; Iwakawa et al, 2011). c-MYC amplification was associated with lymph node metastasis with indefinite meaning for patient survival (Kubokura et al, 2001; Rapp et al, 2009), whereas chromosome 8 gain might be a potential prognostic factor (Kubokura et al, 2001).
Recently, c-MYC amplification was observed as a significant poor-prognostic factor by using whole genome copy number analysis and real-time genomic polymerase chain reaction (RT-G-PCR) in small-sized or early stage lung adenocarcinoma (Iwakawa et al, 2011). From this result, we hypothesised that (1) c-MYC gain might be a useful molecular marker predicting poor prognosis in early stage adenocarcinoma by using fluorescence in situ hybridisation (FISH) method, which is a practical diagnostic tool in the hospital pathology laboratory, and (2) c-MYC gain might have selective utility in lung adenocarcinomas with an activating alteration in EGFR, KRAS, and/or ALK.
In the present study, we intended to verify and validate the clinical applicability of the copy number gain of c-MYC by using FISH in a relatively large-scale cohort. We investigated the relationship between clinicopathologic parameters and c-MYC gain, chromosome 8 gain, and c-MYC amplification, and analysed its clinical significance according to EGFR, KRAS, and ALK alteration status in lung adenocarcinomas, especially in stage I adenocarcinomas.
Materials and methods
Patients and samples
A total of 255 patients with primary lung adenocarcinomas who underwent surgical resection in Seoul National University Bundang Hospital from May 2003 to November 2009 were enroled in this retrospective study. None of them received preoperative chemotherapy or radiation therapy. Clinical and pathologic data were retrieved from patients’ medical records including pathologic reports. Two pathologists (ANS and JHC) reviewed the hematoxylin and eosin-stained slides. The histological subtypes were determined on the basis of the International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society (IASLC/ATS/ERS) classification (Yoshizawa et al, 2011; Warth et al, 2012). Disease-free survival (DFS) was estimated from the date of surgical resection to the date of the initial tumour relapse, and overall survival (OS) was measured from the date of surgery to the time of death. Follow-up period for OS ranged from 1 to 84 months (median OS, 40.0 months), and follow-up period for DFS ranged from 1 to 84 months (median DFS, 29.0 months). The Institutional Review Board (IRB) of the Seoul National University Bundang Hospital approved this study as a study with less than minimal risk, and approved a waiver for consent for this study.
Tissue microarrays (TMA)
The TMA blocks were manufactured from the most representative areas of individual paraffin blocks, as previously described (Superbiochips Laboratories, Seoul, Korea) (Yoo et al, 2010). Briefly, the representative tumour area was selected by two experienced pulmonary pathologists (HK and SBY), and the single core of 2 mm in diameter for each case was taken to TMA block. The cores containing more than 15% of tumour cells by area were considered as valid cores.
Fluorescence in situ hybridisation
To evaluate the copy number of c-MYC and centromeric enumeration probe 8 (CEP8), FISH assay was performed on the TMA sections of 3 μm thickness by using c-MYC probe (Abbott Molecular, Abbott Park, IL, USA) that hybridises to 8q24.12-q24.13 (c-MYC) with Spectrum Orange (red) signal, CEP8 probe (Abbott Molecular) that hybridises the centromeric (alpha satellite) region of chromosome 8 (8p11.1-q11.1) with Spectrum Green signal, and Hybrite (Abbott Molecular), according to manufacturer’s instruction as previously described (Paik et al, 2011).
The FISH slide was interpreted by two experienced pathologists (ANS and JHP) without information about the clinicopathologic characteristics. Tumour tissue was scanned to detect hot spots for c-MYC copy numbers by using × 600 magnification. If the c-MYC signals were homogeneously distributed, then random areas were selected to count the signals. Twenty non-overlapping tumour nuclei from three hot spots or random areas, that is, a total of 60 nuclei, per case were evaluated, and the numbers of c-MYC and CEP8 signals were counted at × 1000 magnification. The 60 nuclei criteria was determined beyond the level of the previous c-MYC gain studies in other solid tumours by Darcy et al (2009) (50 nuclei per case in ovary cancer), and Perez et al (2011) (30 nuclei per case in breast cancer). Small or large clusters of signals were considered as 6 and 12 signals, respectively, according to the interpretive guide for Ventana INFORM HER2 DNA probe staining of breast carcinoma (Ventana Medical Systems, Tucson, AZ, USA). Average copy number of c-MYC and CEP8 per nucleus and their ratio (c-MYC:CEP8) were calculated to determine c-MYC gain, chromosome 8 gain, and c-MYC amplification.
Analysis of EGFR, KRAS mutations, and ALK rearrangement
Epidermal growth factor receptor mutations at exons 18 to 21 and KRAS mutations at codons 12, 13, and 61 were analysed by using PCR and a direct DNA sequencing method with formalin-fixed paraffin-embedded (FFPE) tissue samples, as described previously by Paik et al (2011) and Lee et al (2013). ALK rearrangement was evaluated by using FISH method with ALK probe (Vysis LSI ALK dual-colour, break apart rearrangement probe; Abbott Molecular) with the cutoff value of 15%, as previously described by Paik et al (2011).
Statistical analysis
Statistical analysis was performed by SPSS 18.0 (SPSS Inc, Chicago, IL, USA). To analyse the correlation between clinicopathologic parameters, χ2-test, Fisher’s exact test and Mann–Whitney U-test, and/or Pearson correlation test were used. Receiver-operating characteristic (ROC) curve analysis against 5-year survival was performed to determine the clinically relevant cutoff points of c-MYC and CEP8 copy numbers. For survival analysis, Kaplan–Meier method with log-rank test and multivariate Cox proportional hazards regression analysis were performed. P-values<0.05 were considered as statistically significant (two-tailed). Although all values in this study are from the result of all inclusive enter method where the variables were introduced in one step, the main result was re-tested using other approaches and confirmed that the findings are valid. The influence of EGFR-mutational status on DFS and OS was excluded by testing the effect of EGFR mutation in any multivariate modelling. Post-operative adjuvant therapy had a significant influence on multivariate modelling. Patients directed to receive adjuvant therapy tended to have more advanced stage and an increased risk of recurrence, and current therapies are not very effective in these high-risk patients. As such, post-operative adjuvant therapy was excluded from analyses of DFS and OS.
Results
Clinicopathologic characteristics of patients with total lung adenocarcinomas
The clinicopathologic characteristics of a total of 255 cases of lung adenocarcinoma were summarised in Table 1. Briefly, median age was 64 years with similar frequencies between men (50.6%, 129 out of 255) and women (49.4%, 126 out of 255). Histologically, acinar-predominant type (59.6%, 152 out of 255) was most common. Stage I cases accounted for 59.6% (152 out of 255), while stage II and stage III cases were 14.5 (37 out of 255) and 25.9% (66 out of 255), respectively. Post-operative adjuvant chemotherapy was performed in 39.6% (101 out of 255) and adjuvant radiation therapy in 11.8% (30 out of 255). The clinical profiles of the ‘adjuvant therapy group’ and ‘no adjuvant therapy group’ were not different except for stage (Table 1).
Determining the clinically relevant cutoff value in c-MYC and chromosome 8 gain
The median of c-MYC and CEP8 copy number per nucleus was 1.57 (range, 1.0–22.68) and 1.98 (range: 1.0–5.88). The median ratio (c-MYC:CEP8) were 0.78 (range, 0.32–6.84). Representative FISH patterns were shown in Figure 1. Owing to the lack of established criteria for c-MYC gain, we tested the previously published criteria (Morrison et al, 2005; Darcy et al, 2009; Perez et al, 2011), but clinical significance was limited.

Representative patterns of FISH of c-MYC gene (red colour) and chromosome 8 (CEP8) (green colour) copy number status. (A) High-level gain of c-MYC (c-MYC:CEP8 ratio=6.84), (B) low-level gain of c-MYC (c-MYC:CEP8 ratio=1.82), (C) chromosome 8 gain (c-MYC:CEP8 ratio=0.30), and (D) non-gain of c-MYC or chromosome 8 (c-MYC:CEP8 ratio=0.90).
By ROC curve analysis for predicting 5-year survival (Figure 2), several candidate cutoff values were tested, and the cutoff value representing maximum χ2 (minimum P-value) was selected. ‘c-MYC>2.0 copies per nucleus’ and ‘CEP8⩾3.0 copies per nucleus’ were observed as the most predictive cutoff criteria. In the present study, we defined the ‘c-MYC gain’ as ‘c-MYC>2.0 copies per nucleus’, ‘chromosome 8 gain’ as ‘CEP8⩾3.0 copies per nucleus’, and ‘c-MYC amplification’ as ‘c-MYC:CEP8 ratio⩾2’, respectively. Additionally, to descriptively analyse the characteristics of the c-MYC gain cancer, we arbitrarily subdivided the c-MYC gain (>2.0) into high-level gain (>5.0) and low-level gain (c-MYC copies >2 and ⩽5). By the present criteria, ‘c-MYC gain’ includes specific gain of c-MYC region and chromosome 8 gain.

Scatter plot and ROC curves for c-MYC gain, chromosome 8 gain, and c-MYC amplification (gain of c-MYC :CEP8 ratio) in lung adenocarcinomas. (A) Scatter plot with c-MYC (X-axis) status and chromosome 8 (Y-axis) status. (B) ROC curves for c-MYC:CEP8 ratio, (C) c-MYC gain, and (D) chromosome 8 gain for predicting 5-year survival.
In the full cohort, c-MYC gain was observed in 20.0% (51 out of 255). Low-level gain was observed in 18.4% (47 out of 255) of cancers, and high-level gain was seen in 1.6% (4 out of 255) of cases. Chromosome 8 gain accounted for 5.5% (14 out of 255), and c-MYC amplification was observed only in 2.4% (6 out of 255).
Mutations in EGFR, KRAS, ALK rearrangement, c-MYC gain, chromosome 8 gain, and c-MYC amplification
In full cohort, EGFR and KRAS mutation data were obtained in 94% (239 out of 255) and 48% (123 out of 255) of full cohort. ALK rearrangement data were available in 80% (205 out of 255). As shown in Table 1, EGFR mutation was observed in 49.4% (118 out of 239), KRAS mutation in 5.7% (7 out of 123) and ALK rearrangement in 4.9% (10 out of 205). In two cases, mutations for both EGFR and KRAS were observed. However, ALK rearrangement was not observed in EGFR- or KRAS-mutant cases.
c-MYC gain was observed in 20% (51 out of 255), chromosome 8 gain in 5.5% (14 out of 255), and c-MYC amplification in 2.4% (6 out of 255) (Table 2).
Associations with clinical covariates
Table 2 and Supplementary Table S1-2 show the correlation between c-MYC gain, chromosome 8 gain, c-MYC amplification status and clinical covariates in full cohort (Table 2), stage I (Supplementary Table S1), EGFR wild-type, and EGFR-mutant subsets (Supplementary Table S2). Supplementary Table S3 summarises the significant correlations with clinical covariates. Briefly, c-MYC gain was correlated with lymphatic invasion and recurrence in full cohort and EGFR-mutant subset. Chromosome 8 gain tended to be associated with ever smoker, male sex, and/or lymphatic invasion. But the significance of correlation might be limited owing to relatively small number of cases with chromosome 8 gain (n=14 in full cohort) or c-MYC amplification (n=6 in full cohort).
c-MYC gain was detected in EGFR-mutant cases and EGFR/KRAS/ALK non-mutated cases. Specifically, c-MYC gain was observed in 19% (22 out of 118) of EGFR-mutant cases and 20% (24 out of 121) of EGFR wild-type cases. However, c-MYC gain was not observed in patients with KRAS mutation or ALK rearrangement (Table 2).
Associations with DFS in full cohort
As shown in Table 3, in univariate analysis in full cohort, the conventional clinicopathologic variables, that is pleural invasion, venous invasion, lymphatic invasion, perineural invasion, and high stage (III), were observed as significant poor-prognostic factors for DFS. Both of the c-MYC gain and c-MYC amplification were also poor-prognostic factors for DFS (Figure 3A), while chromosome 8 gain had only marginal significance. In multivariate analysis with c-MYC gain and conventional significant variables, c-MYC gain was a significant prognostic factor for DFS (P=0.022; HR=1.71), while c-MYC amplification was not significant (P=0.589) when c-MYC amplification was included instead of c-MYC gain in the modelling. Although adjuvant therapy was another influential factor for DFS and OS (P<0.001 for both), we did not include it in the multivariate model of Table 3, because ‘adjuvant therapy group’ showed significantly high rate of recurrence and poor prognosis (13.3% (19 out of 143) of recurrences in no adjuvant therapy subgroup; 73.2% (82 out of 112) of recurrence in adjuvant therapy subgroup; P<0.001). This might suggest that the adjuvant therapy had the role of clinical surrogate marker of high probability of recurrence overriding its innate therapeutic effects. Consistent with this, by including adjuvant therapy in the multivariate modelling, the significance of c-MYC gain became limited (P=0.080), while the inclusion of EGFR mutation did not significantly influence on DFS using univariate and multivariate models (data not shown).

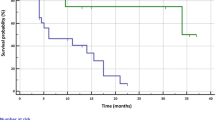
Kaplan–Meier survival curves illustrating prognostic effects of c-MYC gain in lung adenocarcinomas. (A) c-MYC gain for DFS and (B) OS in full cohort. (C) c-MYC gain for DFS in stage I, (D) EGFR-mutant, and (E) EGFR-mutant low stage (I, II) subgroups. (F) c-MYC gain for OS in stage I subgroup.
Associations with OS in full cohort
In univariate analysis for OS, the c-MYC gain and chromosome 8 gain were significant prognostic factors, along with conventional clinicopathologic variables (Table 3 and Figure 3B). In multivariate analysis for OS, c-MYC gain, high stage (III), pleural, and venous invasions were independent poor-prognostic factors. High stage (III) was the most powerful prognostic factor (P<0.001, HR=4.88), while c-MYC gain was a less powerful significant prognostic factor (P=0.032, HR=2.04). The multivariate modelling was not influenced by inclusion of adjuvant therapy or EGFR mutation (data not shown).
Subset analysis of DFS
To investigate the clinical and prognostic significance of c-MYC gain in several clinicopathologic subgroups, we analysed the survival effect of MYC gain in stage I subgroup, EGFR-mutant and wild-type subgroups.
As shown by Iwakawa et al (2011), we performed survival analysis in the stage I adenocarcinoma subgroup containing 152 patients. In this subgroup, c-MYC gain was observed in 15.8% (24 out of 152), chromosome 8 gain in 4.6% (7 out of 152), and c-MYC amplification in 1.3% (2 out of 152). In univariate survival analysis of stage I adenocarcinoma subgroup, c-MYC gain tended to show poor prognosis but the association with DFS did not achieve statistical significance (P=0.065, Table 4 and Figure 3C). In multivariate analysis with c-MYC gain and conventional variables, c-MYC gain was the independent prognostic factor for DFS in stage I subgroup (P=0.023, HR=4.70; Table 4; Figure 4A).

Forest plot showing multivariate survival analysis for DFS and OS in stage I, EGFR -mutant, and EGFR wild-type subgroups. (A) DFS in stage I, (B) EGFR-mutant, and (C) EGFR wild-type subgroups. (D) OS in stage I, (E) EGFR-mutant, and (F) EGFR wild-type subgroups.
Next, we investigated the clinical meaning of c-MYC gain according to EGFR-mutational status, since c-MYC gain was frequently observed in lung adenocarcinoma with EGFR-mutation (Table 2). The frequency of c-MYC gain was similar between the two subgroups (19% (22 out of 118) of EGFR-mutant subgroup and 20% (24 out of 121) of EGFR wild-type subgroup) (Table 2 and Supplementary Table S2).
In patients with a mutation in EGFR, c-MYC gain was associated with DFS using univariate analysis (P=0.008; Figure 3D and E) and exhibited independent prognostic relevance in multivariate analysis (P=0.022) after adjusting for the conventional clinical covariates (Table 5 and Figure 4B). In patients without EGFR mutation, c-MYC gain, chromosome 8 gain, and c-MYC amplification were not associated with DFS (Table 5 and Figure 4C).
Subset analysis of OS
In stage I subgroup, c-MYC gain was observed as a significant poor-prognostic factor for OS (P=0.008; Table 4 and Figure 3F), while chromosome 8 gain and c-MYC amplification were not significant (Table 4) in univariate analysis. In multivariate analysis with c-MYC gain and significant conventional variables, c-MYC gain was the independent poor-prognostic factor for OS (P=0.031, HR=4.65, Table 4 and Figure 4D).
In patients with a mutation in EGFR, c-MYC gain, chromosome 8 gain, and c-MYC amplification were not associated with OS (Table 5 and Figure 4E). In patients without EGFR mutation, high stage (III) (but not c-MYC gain or chromosome 8 gain) was a significant prognostic factor for OS in multivariate analysis (P<0.001, Table 5 and Figure 4F).
Discussion
In the present retrospective study with FISH method, we investigated the clinicopathologic significance of c-MYC gain, chromosome 8 gain, and c-MYC amplification in lung adenocarcinomas according to the genetic alteration status of EGFR, KRAS, and ALK, and validated the PCR-based results by Iwakawa et al (2011) in early stage adenocarcinoma. Herein, we observed c-MYC gain was an independent poor-prognostic factor for DFS and OS in lung adenocarcinomas, both in full cohort and stage I cancer, and possibly in EGFR-mutant adenocarcinomas for DFS.
c-MYC is known to link the stimulation by growth factors and cellular proliferation in normal cells, and pathologic activation through translocation or amplification is thought to constitutively enhance the transcription of a certain group of genes contributing to cell proliferation without the stimulation by growth factors (Lin et al, 2012). The extremely high-proliferative activity in Burkitt lymphoma is mediated via t(8;14) translocation where c-MYC gene is juxtapositioned with one of the immunoglobulin loci, and regulated by immunoglobulin gene regulatory elements (Boxer and Dang, 2001). The detection of c-MYC translocation by using FISH has been established as a hallmark for the diagnosis or prognosis in malignant lymphoma (Swerdllow et al, 2008; Slack and Gascoyne, 2011). Similarly, c-MYC gain detected by FISH method in some solid tumours tended to be associated with poor-clinical outcome (Sauter et al, 1995; Ghadimi et al, 2003; Morrison et al, 2005; Darcy et al, 2009; Perez et al, 2011). However, the clinical application of c-MYC gain as a prognostic marker in lung cancer has been limited owing to lack of solid evidence (Kubokura et al, 2001). Recently, the Iwakawa’s study using RT-G-PCR method showed c-MYC gain as a significant prognostic marker in early stage lung adenocarcinomas (Iwakawa et al, 2011). By using FISH method, we observed the prognostic significance of c-MYC gain in lung adenocarcinomas.
In the present study, low-level gain of c-MYC (18.4%) was more common than high-level gain (1.6%) in lung adenocarcinoma. Despite the slightly different criteria, similar ‘low-level gain’-predominant patterns have been described in other solid tumours (Sauter et al, 1995; Perez et al, 2011; Zitterbart et al, 2011). Therefore, the copy number analysis of 8q24 or c-MYC locus might be very delicate, and inevitably raises important issues about how to define the ‘c-MYC gain/amplification’ and whether to include the ‘low-level gain’ in the ‘c-MYC gain’ or not. In a recent study with prostate cancer, the relatively ‘low-level gain’ was included in the ‘c-MYC gain/amplification’ group with the criteria of c-MYC/CEP8 ratio >1.5, and a poor prognosis was observed in this group (Fromont et al, 2013). Another recent study with lymphoma, the cases with 2.2 copies per nucleus (44 copies/20 cells) were classified as the increased c-MYC copy number group, and c-MYC protein expression was correlated with increased c-MYC copy number and mRNA expression (Valentino et al, 2013). In agreement with these results, ‘c-MYC gain’ criteria by Iwakawa et al (2011) with lung cancer by using RT-G-PCR was set as low as c-MYC copy number >1.59, while the criteria might not be directly applied to the FISH method. The ‘c-MYC gain’ criteria in the present study (c-MYC>2.0 copies/nucleus) appears to largely contain duplication of 8q24 or c-MYC locus and/or chromosome 8 polysomy, as shown in Figure 2. In the clinical point of view, the relatively simple criteria in the present study with FFPE tissue-based FISH method might be practically useful in the assessment of patients with lung adenocarcinomas, because it could clearly distinguish the poor-prognostic group, especially in stage I cancer, regardless of c-MYC:CEP8 ratio or chromosome 8 status.
It might not be clear how the ‘low-level gain’ of c-MYC gene in the present study could lead to the aggressive biology of the lung cancer, and it is still poorly understood whether the prognostic effect of c-MYC gain is achieved solely by the increased expression of c-MYC protein, since the c-MYC gene status but not c-MYC protein expression did influence the prognosis in a recent prostate cancer study (Fromont et al, 2013). Considering the complex way of regulation of c-MYC protein and multiple interaction with other molecules, it might be possible that the gain of 8q24 or c-MYC locus could enhance c-MYC activity at certain level, which might be sufficient to effectively trigger amplification of transcription involving a various set of genes in tumour cells (Lin et al, 2012), which remains to be investigated further. Additionally, other factors including non-coding RNAs and some regulatory proteins encoded at 8q24 locus or nearby chromosomal regions might be involved in the ‘c-MYC gain’-associated cellular changes leading to poor-clinical outcome. Especially, several candidate protein-coding and non-coding oncogenes including ATAD2, PVT1, and MIR1204, have been known to be mapped in 8q24 locus, coamplified with c-MYC, and transduce or modify the c-MYC-induced signals to other regulatory pathways (Carramusa et al, 2007; Huppi et al, 2008, 2012; Raeder et al, 2013), while the level of functional contribution of these molecules in the tumour biology of lung adenocarcinoma needs to be clarified. In this context, to evaluate the ‘c-MYC gain’-associated phenotypic changes at either cellular or clinical levels, it might not simply be sufficient to investigate the immunohistochemical expression of c-MYC protein, which harbours various epitopes, short half-life, complex interaction with other protein factors, and still unclear significance of various subcellular localisation including cytoplasmic cleavage product (Conacci-Sorrell et al, 2010; Fromont et al, 2013).
The solid tumours with ‘c-MYC gain’ have tended to be associated with invasiveness and lymph node metastasis (Ghadimi et al, 2003; Heselmeyer-Haddad et al, 2012), which are required phenotypes for the localised early cancers to progress and eventually disseminate. Moreover, the c-MYC has been known to be a downstream target of EGFR/RAS/RAF/MEK/ERK signalling pathway (Dang, 2012). c-MYC played an important role in tumour progression in RAF- or RAS-driven lung cancer models in vitro or in vivo, and targeting c-MYC effectively suppressed or reversed tumorigenesis (Tran et al, 2008; Rapp et al, 2009; Fukazawa et al, 2010; Soucek et al, 2013). These findings suggest that c-MYC gain might be involved in the progression of early stage lung adenocarcinoma, especially in conjunction with EGFR/RAS/RAF pathway, and be consistent with the observations in the present study that c-MYC gain was a significant prognostic factor in stage I adenocarcinoma subgroup, as well as in EGFR-mutant subgroup. The meaning of c-MYC gain according to KRAS mutation status could not be sufficiently analysed owing to low KRAS mutation rate, and it remains to be clarified further.
In addition to c-MYC gain, chromosome 8 gain also harboured a poor-prognostic effect in lung adenocarcinomas, while the significance was limited. It might be assumed that the ‘c-MYC gain’ defined in the present study would technically contain a broad spectrum of genomic gains involving c-MYC (8q24) locus, up to some proportion of chromosome 8 gains (Figure 2). Considering that chromosomal regions around 8q24 are unstable (Huppi et al, 2008), our observations from clinical samples suggest that whether or not c-MYC (8q24) region is included in the process of genomic gain involving some parts of chromosome 8 might be a surrogate marker for genomic instability and critical determinant of poor-clinical outcome in lung adenocarcinomas. Conversely, stable genomic status at c-MYC (8q24) locus or nearby regions might indirectly reflect that one of the vulnerable sites of genomic instability associated with the clinical outcome was spared.
As a tissue biomarker of lung adenocarcinoma, c-MYC gain might be a good candidate, because it accounts for 15–20% of full cohort or subsets (Supplementary Table S3), and shows an independent prognostic value in full cohort and stage I cancer. In contrast, the cases with chromosome 8 gain and c-MYC amplification were observed in less than 6% and 3% of full cohort and subsets (Supplementary Table S3), respectively, and did not show any significant prognostic value. Therefore, the clinical meaning of c-MYC gain needs to be validated for clinical application in prospective and larger scale study.
In conclusion, we observed that c-MYC gain was associated with lymphatic invasion, and was an independent poor-prognostic factor for DFS and OS in lung adenocarcinomas, both in full cohort and stage I subgroup, and possibly for DFS in EGFR-mutant subgroup. These findings might provide the useful way of detailed risk stratification in patients with lung adenocarcinomas, and an insight into pathogenesis and mechanism of progression in lung adenocarcinomas. Furthermore, patients with lung adenocarcinoma with c-MYC gain, even in early stage, appear to have an increased risk of disease progression and death, which merits further prospective evaluation across multiple institutions to validate the clinical utility of c-MYC gains in this disease setting and to determine whether these patients might benefit from additional first-line therapy.
Change history
27 May 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arrieta Rodriguez OG, Villarreal-Garza C, Martinez-Barrera L, Morales M, Dorantes-Gallareta Y, Pena-Curiel O, Contreras-Reyes S, Macedo-Perez EO, Alatorre-Alexander J (2013) Usefulness of Serum Carcinoembryonic Antigen (CEA) in evaluating response to chemotherapy in patients with advanced non small-cell lung cancer: a prospective cohort study. BMC Cancer 13: 254.
Boxer LM, Dang CV (2001) Translocations involving c-myc and c-myc function. Oncogene 20: 5595–5610.
Brueckl WM, Al-Batran SE, Ficker JH, Claas S, Atmaca A, Hartmann A, Rieker RJ, Wirtz RM (2013) Prognostic and predictive value of estrogen receptor 1 expression in completely resected non-small cell lung cancer. Int J Cancer 133: 1825–1831.
Carramusa L, Contino F, Ferro A, Minafra L, Perconti G, Giallongo A, Feo S (2007) The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. J Cell Physiol 213: 511–518.
Chen Y, Min L, Zhang X, Hu S, Wang B, Liu W, Wang R, Gu X, Shen W, Lv H, Zou J, Chen Y, Xu X, Chen L (2013) Decreased miRNA-148a is associated with lymph node metastasis and poor clinical outcomes and functions as a suppressor of tumour metastasis in non-small cell lung cancer. Oncol Rep 30 (4): 1832–1840.
Conacci-Sorrell M, Ngouenet C, Eisenman RN (2010) Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 142: 480–493.
Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35.
Darcy KM, Brady WE, Blancato JK, Dickson RB, Hoskins WJ, McGuire WP, Birrer MJ (2009) Prognostic relevance of c-MYC gene amplification and polysomy for chromosome 8 in suboptimally-resected, advanced stage epithelial ovarian cancers: a Gynecologic Oncology Group study. Gynecol Oncol 114: 472–479.
Fromont G, Godet J, Peyret A, Irani J, Celhay O, Rozet F, Cathelineau X, Cussenot O (2013) 8q24 amplification is associated with Myc expression and prostate cancer progression and is an independent predictor of recurrence after radical prostatectomy. Hum Pathol 44 (8): 1617–1623.
Fukazawa T, Maeda Y, Matsuoka J, Yamatsuji T, Shigemitsu K, Morita I, Faiola F, Durbin ML, Soucek L, Naomoto Y (2010) Inhibition of Myc effectively targets KRAS mutation-positive lung cancer expressing high levels of Myc. Anticancer Res 30: 4193–4200.
Ghadimi BM, Grade M, Liersch T, Langer C, Siemer A, Fuzesi L, Becker H (2003) Gain of chromosome 8q23-24 is a predictive marker for lymph node positivity in colorectal cancer. Clin Cancer Res 9: 1808–1814.
Hermeking H (2003) The MYC oncogene as a cancer drug target. Curr Cancer Drug Targets 3: 163–175.
Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, Ortiz-Melendez C, Lee WJ, Christensen R, Prindiville SA, Calzone KA, Soballe PW, Hu Y, Chowdhury SA, Schwartz R, Schaffer AA, Ried T (2012) Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumour heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol 181: 1807–1822.
Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ (2012) The 8q24 gene desert: an oasis of non-coding transcriptional activity. Front Genet 3: 69.
Huppi K, Volfovsky N, Runfola T, Jones TL, Mackiewicz M, Martin SE, Mushinski JF, Stephens R, Caplen NJ (2008) The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol Cancer Res 6: 212–221.
Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansen S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Janne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M (2012) Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120.
Iwakawa R, Kohno T, Kato M, Shiraishi K, Tsuta K, Noguchi M, Ogawa S, Yokota J (2011) MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clin Cancer Res 17: 1481–1489.
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60: 277–300.
Job B, Bernheim A, Beau-Faller M, Camilleri-Broet S, Girard P, Hofman P, Mazieres J, Toujani S, Lacroix L, Laffaire J, Dessen P, Fouret P (2010) Genomic aberrations in lung adenocarcinoma in never smokers. PLoS One 5: e15145.
Johnson BE, Ihde DC, Makuch RW, Gazdar AF, Carney DN, Oie H, Russell E, Nau MM, Minna JD (1987) myc family oncogene amplification in tumour cell lines established from small cell lung cancer patients and its relationship to clinical status and course. J Clin Invest 79: 1629–1634.
Kawase A, Yoshida J, Miyaoka E, Asamura H, Fujii Y, Nakanishi Y, Eguchi K, Mori M, Sawabata N, Okumura M, Yokoi K (2013) Visceral pleural invasion classification in non-small-cell lung cancer in the 7th edition of the tumour, node, metastasis classification for lung cancer: validation analysis based on a large-scale nationwide database. J Thorac Oncol 8: 606–611.
Kim H, Yoo SB, Sun P, Jin Y, Jheon S, Lee CT, Chung J-H (2013a) Alteration of the E-Cadherin/β-Catenin Complex Is an Independent Poor Prognostic Factor in Lung Adenocarcinoma. Korean J Pathol 47: 44–51.
Kim HS, Mitsudomi T, Soo RA, Cho BC (2013b) Personalised therapy on the horizon for squamous cell carcinoma of the lung. Lung Cancer 80: 249–255.
Kubokura H, Tenjin T, Akiyama H, Koizumi K, Nishimura H, Yamamoto M, Tanaka S (2001) Relations of the c-myc gene and chromosome 8 in non-small cell lung cancer: analysis by fluorescence in situ hybridisation. Ann Thorac Cardiovasc Surg 7: 197–203.
Kudo Y, Saji H, Shimada Y, Nomura M, Matsubayashi J, Nagao T, Kakihana M, Usuda J, Kajiwara N, Ohira T, Ikeda N (2012) Impact of visceral pleural invasion on the survival of patients with non-small cell lung cancer. Lung Cancer 78: 153–160.
Kwon MJ, Seo J, Kim YJ, Kwon MJ, Choi JY, Kim TE, Lee DH, Park S, Shin YK, Han J, Choi YL (2013) Prognostic significance of CD151 overexpression in non-small cell lung cancer. Lung Cancer 81: 109–116.
Lee HJ, Xu X, Kim H, Jin Y, Sun P, Kim JE, Chung J-H (2013) Comparison of Direct Sequencing, PNA Clamping-Real Time Polymerase Chain Reaction, and Pyrosequencing Methods for the Detection of EGFR Mutations in Non-small Cell Lung Carcinoma and the Correlation with Clinical Responses to EGFR Tyrosine Kinase Inhibitor Treatment. Korean J Pathol 47: 52–60.
Li Z, Owonikoko TK, Sun SY, Ramalingam SS, Doetsch PW, Xiao ZQ, Khuri FR, Curran WJ, Deng X (2012) c-Myc suppression of DNA double-strand break repair. Neoplasia 14: 1190–1202.
Liao DJ, Dickson RB (2000) c-Myc in breast cancer. Endocr Relat Cancer 7: 143–164.
Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151: 56–67.
Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD (1983) Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306: 194–196.
Mok TS (2011) Personalized medicine in lung cancer: what we need to know. Nat Rev Clin Oncol 8: 661–668.
Morrison C, Radmacher M, Mohammed N, Suster D, Auer H, Jones S, Riggenbach J, Kelbick N, Bos G, Mayerson J (2005) MYC amplification and polysomy 8 in chondrosarcoma: array comparative genomic hybridization, fluorescent in situ hybridization, and association with outcome. J Clin Oncol 23: 9369–9376.
Nentwich MF, Bohn BA, Uzunoglu FG, Reeh M, Quaas A, Grob TJ, Perez D, Kutup A, Bockhorn M, Izbicki JR, Vashist YK (2013) Lymphatic invasion predicts survival in patients with early node-negative non-small cell lung cancer. J Thorac Cardiovasc Surg. 146 (4): 781–787.
Paik JH, Choe G, Kim H, Choe JY, Lee HJ, Lee CT, Lee JS, Jheon S, Chung JH (2011) Screening of anaplastic lymphoma kinase rearrangement by immunohistochemistry in non-small cell lung cancer: correlation with fluorescence in situ hybridisation. J Thorac Oncol 6: 466–472.
Pao W, Girard N (2011) New driver mutations in non-small-cell lung cancer. Lancet Oncol 12: 175–180.
Perez EA, Jenkins RB, Dueck AC, Wiktor AE, Bedroske PP, Anderson SK, Ketterling RP, Sukov WR, Kanehira K, Chen B, Geiger XJ, Andorfer CA, McCullough AE, Davidson NE, Martino S, Sledge GW, Kaufman PA, Kutteh LA, Gralow JR, Harris LN, Ingle JN, Lingle WL, Reinholz MM (2011) C-MYC alterations and association with patient outcome in early-stage HER2-positive breast cancer from the north central cancer treatment group N9831 adjuvant trastuzumab trial. J Clin Oncol 29: 651–659.
Raeder MB, Birkeland E, Trovik J, Krakstad C, Shehata S, Schumacher S, Zack TI, Krohn A, Werner HM, Moody SE, Wik E, Stefansson IM, Holst F, Oyan AM, Tamayo P, Mesirov JP, Kalland KH, Akslen LA, Simon R, Beroukhim R, Salvesen HB (2013) Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS One 8: e54873.
Rapp UR, Korn C, Ceteci F, Karreman C, Luetkenhaus K, Serafin V, Zanucco E, Castro I, Potapenko T (2009) MYC is a metastasis gene for non-small-cell lung cancer. PLoS One 4: e6029.
Sauter G, Carroll P, Moch H, Kallioniemi A, Kerschmann R, Narayan P, Mihatsch MJ, Waldman FM (1995) c-myc copy number gains in bladder cancer detected by fluorescence in situ hybridisation. Am J Pathol 146: 1131–1139.
Sawabata N, Asamura H, Goya T, Mori M, Nakanishi Y, Eguchi K, Koshiishi Y, Okumura M, Miyaoka E, Fujii Y (2010) Japanese Lung Cancer Registry Study: first prospective enrollment of a large number of surgical and nonsurgical cases in 2002. J Thorac Oncol 5: 1369–1375.
Shimizu K, Funai K, Sugimura H, Sekihara K, Kawase A, Shiiya N (2013) D2-40-positive lymphatic vessel invasion is not a poor prognostic factor in stage I lung adenocarcinoma. Pathol Int 63: 201–205.
Slack GW, Gascoyne RD (2011) MYC and aggressive B-cell lymphomas. Adv Anat Pathol 18: 219–228.
Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, Evan GI (2013) Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 27: 504–513.
Sun PL, Jin Y, Kim H, Seo AN, Jheon S, Lee CT, Chung JH (2013) Survivin expression is an independent poor prognostic marker in lung adenocarcinoma but not in squamous cell carcinoma. Virchows Arch 463 (3): 427–436.
Swerdllow S, Campo E, Harris NL (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. IARC.
Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J, Horng G, Bellovin DI, Giuriato S, Wang CS, Whitsett JA, Felsher DW (2008) Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One 3: e2125.
Valentino C, Kendrick S, Johnson N, Gascoyne R, Chan WC, Weisenburger D, Braziel R, Cook JR, Tubbs R, Campo E, Rosenwald A, Ott G, Delabie J, Jaffe E, Zhang W, Brunhoeber P, Nitta H, Grogan T, Rimsza L (2013) Colorimetric in situ hybridisation identifies MYC gene signal clusters correlating with increased copy number, mRNA, and protein in diffuse large B-cell lymphoma. Am J Clin Pathol 139: 242–254.
Warth A, Muley T, Meister M, Stenzinger A, Thomas M, Schirmacher P, Schnabel PA, Budczies J, Hoffmann H, Weichert W (2012) The novel histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification system of lung adenocarcinoma is a stage-independent predictor of survival. J Clin Oncol 30: 1438–1446.
Woo T, Okudela K, Mitsui H, Tajiri M, Yamamoto T, Rino Y, Ohashi K, Masuda M (2012) Prognostic value of the IASLC/ATS/ERS classification of lung adenocarcinoma in stage I disease of Japanese cases. Pathol Int 62: 785–791.
Yoo SB, Lee HJ, Park JO, Choe G, Chung DH, Seo JW, Chung JH (2010) Reliability of chromogenic in situ hybridisation for epidermal growth factor receptor gene copy number detection in non-small-cell lung carcinomas: a comparison with fluorescence in situ hybridisation study. Lung Cancer 67: 301–305.
Yoshizawa A, Motoi N, Riely GJ, Sima CS, Gerald WL, Kris MG, Park BJ, Rusch VW, Travis WD (2011) Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: prognostic subgroups and implications for further revision of staging based on analysis of 514 stage I cases. Mod Pathol 24: 653–664.
Zhang L, Hou Y, Ashktorab H, Gao L, Xu Y, Wu K, Zhai J (2010) The impact of C-MYC gene expression on gastric cancer cell. Mol Cell Biochem 344: 125–135.
Zitterbart K, Filkova H, Tomasikova L, Necesalova E, Zambo I, Kantorova D, Slamova I, Vranova V, Zezulkova D, Pesakova M, Pavelka Z, Veselska R, Kuglik P, Sterba J (2011) Low-level copy number changes of MYC genes have a prognostic impact in medulloblastoma. J Neurooncol 102: 25–33.
Acknowledgements
This research was supported by Basic Science Research Programme through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2012R1A1A2006898 to JHP), Republic of Korea, and partly supported by a Grant-in-Aid from the Korea Healthcare technology R&D project, Ministry of Health and Welfare, Republic of Korea (A111405 to JHC).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Seo, A., Yang, J., Kim, H. et al. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. Br J Cancer 110, 2688–2699 (2014). https://doi.org/10.1038/bjc.2014.218
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.218
Keywords
This article is cited by
-
Assessment of MYC and TERT copy number variations in lung cancer using digital PCR
BMC Research Notes (2023)
-
Chemoresistance Mechanisms in Non-Small Cell Lung Cancer—Opportunities for Drug Repurposing
Applied Biochemistry and Biotechnology (2023)
-
A novel TRPM7/O-GlcNAc axis mediates tumour cell motility and metastasis by stabilising c-Myc and caveolin-1 in lung carcinoma
British Journal of Cancer (2020)
-
Expression and clinical significance of PD-L1 and c-Myc in non-small cell lung cancer
Journal of Cancer Research and Clinical Oncology (2019)
-
PIG3 promotes NSCLC cell mitotic progression and is associated with poor prognosis of NSCLC patients
Journal of Experimental & Clinical Cancer Research (2017)
