
Abstract
Trabectedin is the first marine-derived anti-neoplastic drug approved for the treatment of advanced soft tissue sarcoma and, in combination with pegylated liposomal doxorubicin, for the treatment of patients with relapsed platinum-sensitive ovarian cancer. From the beginning of its development, trabectedin showed some peculiar properties that clearly distinguished it from other anti-cancer drugs. In this mini-review, we will outline the current state of knowledge regarding the mode of action of trabectedin, which appears to represent a new class of anti-neoplastic drugs acting both on cancer cells and on the tumour microenvironment.
Similar content being viewed by others

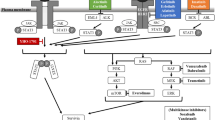
Main
Trabectedin is a tetrahydroisoquinoline alkaloid that was initially isolated from the Caribbean tunicate Ecteinascida turbinata and currently prepared by chemical synthesis (Figure 1A) (Cuevas and Francesch, 2009). It was previously described that trabectedin binds to specific selected triplets of the DNA minor groove, whereas a part of it protrudes out of the DNA and probably interacts with proteins at the site of adduct such as XPG or RNA polymerase II (Pol II) (Figure 1B) (Hurley and Zewail-Foote, 2001; Gago and Hurley, 2002; Herrero et al, 2006; D’Incalci and Galmarini, 2010; Feuerhahn et al, 2011). Besides these molecular mechanisms, trabectedin also induces unexpected biological effects; for example, it is active at low nM concentrations whereas DNA-interacting agents (e.g., temozolomide or platinum compounds) act in the high μ M range. It is also worthwhile noting that trabectedin affects slow growing cells and cells in the G1 phase of the cell cycle; furthermore, the pattern of sensitivity observed in cells deficient in DNA repair mechanisms is different. For example, cells deficient in nucleotide excision repair (NER) are generally more sensitive to cisplatin while they are partially resistant to trabectedin (Damia et al, 2001; Erba et al, 2001; Takebayashi et al, 2001; Damia and D’Incalci, 2007). On the other hand, cells deficient in homologous recombination (HR) (e.g., with mutations of BRCA1 or BRCA2 genes) are sensitive to trabectedin as well as to platinum compounds (Damia and D’Incalci, 2007; Soares et al, 2007; Tavecchio et al, 2008).

(A) Chemical structure of trabectedin; (B) trabectedin binding to the DNA minor groove. The figure illustrates that a part of the molecule binds to DNA whereas another part protrudes out of DNA.
Inhibition of trans-activated transcription
What appears unique for trabectedin is that, at pharmacological concentrations, it modulates gene expression in a promoter- and gene-dependent manner. This finding, initially demonstrated for the promoters of heat-shock proteins (HSPs) and MDR1 genes, was subsequently extended to several other promoters and appears to be cell dependent (Jin et al, 2000; Minuzzo et al, 2000; Feuerhahn et al, 2011). Moreover, it was reported that trabectedin affects trans-activated but not basal transcription using different mechanisms (Friedman et al, 2002). First, trabectedin inhibits transcription by preventing the binding of transcription factors to DNA (see below) (Forni et al, 2009; Di Giandomenico et al, 2013). Second, the adducts formed by trabectedin functionally mimic an inter-strand cross-linking lesion, despite the fact that the drug binds covalently to only one DNA strand (its interaction with the opposite strand being only through van der Waals forces and hydrogen bonds). This type of lesion is highly effective in blocking transcription through stabilisation of the ds-DNA (Bueren-Calabuig et al, 2011; Feuerhahn et al, 2011). Finally, trabectedin interacts directly with the elongating Pol II regardless of the strand they are located on (Feuerhahn et al, 2011). This eventually arrests Pol II during transcription elongation and induces its rapid and massive degradation via the proteasome pathway in a transcription coupled NER-dependent manner (Aune et al, 2008). Therefore, it seems that the activity of trabectedin is mainly related to direct effects on transcription regulation.
Specific mechanisms against some translocation-related sarcomas
Myxoid liposarcomas are mainly associated with the presence of t(12;16) or t(12;22) translocations that fuses the gene CHOP (at 12q13.1–q13.2) with FUS (at 16p11.2) or EWS (at 22q12.2), respectively. The fusion proteins FUS-CHOP or EWS-CHOP act as abnormal transcription factors and are involved in the pathogenesis of the disease. Recent experiments have demonstrated that trabectedin blocks the trans-activating ability of these chimaeras by displacing the oncogenic fusion protein FUS-CHOP from its target promoters (Forni et al, 2009; Di Giandomenico et al, 2013). This eventually induces adipogenic differentiation of myxoid liposarcoma cells. This effect has been observed in FUS-CHOP expressing experimental models (tumour cells, myxoid liposarcoma xenografts and transgenic models) and in tumour biopsies taken before and after trabectedin therapy in patients with myxoid liposarcoma (Charytonowicz et al, 2012; Di Giandomenico et al, 2013). It was also found that, while the displacement of FUS-CHOP chimaera variants (e.g., subtypes I, II and III) from target promoters is similar, the kinetic of re-attachment of the fusion protein subtypes appears to differ. In fact, re-attachment is faster for type III than for types I and II. This is possibly due to the larger size of type III chimaera that makes the complex of transcription factor cofactors and DNA more stable. Of note, type III myxoid liposarcoma experimental models appear to be less sensitive to the differentiation and anti-angiogenic effects of trabectedin, suggesting that the inhibition time of the chimaeric protein is apparently important for its pharmacological effects. It is also interesting to note that FUS-CHOP expression influences several genes involved in tumour angiogenesis and cancer inflammation (Germano et al, 2010). Therefore, the peculiar activity of trabectedin in myxoid liposarcomas (Grosso et al, 2007, 2009) could be related to an effect not only on genes involved in adipocytic differentiation, but also on genes that code for factors which affect the tumour microenvironment (TME).
Modulation of transcription by trabectedin has also been shown in Ewing’s sarcoma, a neoplasm driven by the oncogenic fusion gene EWS-FLI1 (Grohar et al, 2011; Grohar and Helman, 2013). Recently, it has been shown that this modulation can be exploited to select a very effective combination treatment of trabectedin followed by irinotecan (Grohar et al, 2013). The basis of this synergism is that, by blocking the trans-activating ability of EWS-FLI1 protein, trabectedin induces a downregulation of Werner Syndrome protein (WRN) that increases susceptibility to topoisomerase I poisons (Grohar et al, 2011; Grohar and Helman, 2013). The sequential use of trabectedin and irinotecan has been found to be very effective not only in vitro but also in tumour xenografts derived from Ewing’s sarcoma patients (Grohar et al, 2011; Grohar and Helman, 2013). These results may provide the rational for clinical studies on the sequential combination of trabectedin and irinotecan in Ewing’s sarcoma, a tumour for which there is an urgent and unmet need for more effective therapies.
Effects of trabectedin on the TME
Since the last decade, tumours are considered as complex tissues with a dynamic and reactive TME. This TME is populated by different non-neoplastic cells (e.g., inflammatory leukocytes, activated fibroblasts and endothelial cells) that actively communicate with cancer cells via chemokines or cytokines. It is now well established that the persistence of inflammatory pathways in the TME is linked with tumour promotion (Bissell and Hines, 2011). Among stromal cells, tumour-associated macrophages (TAMs), derived from blood circulating monocytes and functionally ‘educated’ by tumour cells, display a number of pro-tumoral functions, such as production of growth factors, switch-on of neo-angiogenesis, enhanced protease activity and support in tumour cell dissemination (Mantovani et al, 2002; Pollard, 2004; Qian and Pollard, 2010; Allavena and Mantovani, 2012). It was therefore of interest to find that, in a tumour, trabectedin was affecting not only the neoplastic compartment but also monocytes and macrophages, which are quiescent cells and therefore considered as poorly sensitive to classical DNA-damaging agents (Galmarini et al, 2014). These findings opened an important area of research for the implications of macrophage pro-tumoral functions and disease progression.
The exquisite selectivity of trabectedin against mononuclear phagocytes (monocytes and macrophages) was recently related to the rapid activation of caspase 8 by membrane signalling TRAIL receptors; these receptors are expressed more in monocytes/macrophages than in neutrophils and lymphocytes (Germano et al, 2013). It was also shown that short exposure to trabectedin caused a strong decrease in the production of several cytokines and chemokines secreted by monocytes/macrophages and tumour cells. For example, trabectedin treatment downregulates the expression of IL-6, CCL2, CXCL8, Angiopoietin 2 or VEGF, but not of other biological mediators such as TNFα (Allavena et al, 2005; Germano et al, 2010). The anti-inflammatory effects of trabectedin were confirmed in different tumour xenografts and in human soft tissue sarcoma samples from patients receiving trabectedin as neo-adjuvant therapy (Germano et al, 2013). The importance of this mechanism of anti-cancer activity was definitely proven in experiments where tumour cells resistant to trabectedin in vitro were injected into mice. Interestingly, in the in vivo setting, trabectedin still showed anti-tumour activity (Germano et al, 2013). The results clearly indicated that the macrophage-targeted effect was sufficient to significantly reduce neoplastic growth, under conditions where tumour cells were unresponsive to the drug. Thus, trabectedin effects on the TME and on macrophages are—at least in part—important contributors of its anti-tumour and anti-metastatic activity. A summary of these effects are depicted in Figure 2.

Schematic summary of the different mechanisms of action and biological effects on cancer cells and tumour microenvironment.
Clinical implications and open questions
According to the above findings, trabectedin not only inhibits the growth of cancer cells but also affects the TME by limiting the numbers of macrophages and by inhibiting the production of macrophage products that eventually promote tumour growth. In reference to its activity on neoplastic cells, the ability of trabectedin to modulate oncogenic transcription factors is certainly very appealing for several tumours whose pathogenesis is related to translocations involving transcription factor activity. This is the case for some soft tissue sarcoma, but the concept could be extended to other solid tumours and haematological malignancies. To our knowledge, trabectedin is the first compound able to displace an oncogenic transcription factor from its target promoters with high specificity. Unpublished data from our laboratories, however, suggest that in some cases (e.g., in Ewing’s sarcoma) the inhibition of transcription factor binding to target sequences does not last very long, and transcription modulation occurs for a short time, possibly not sufficient to exert a significant anti-tumour effect. Nevertheless, a transient change in the transcription of some genes can be exploited to identify appropriate combinations, as recently highlighted in the case of Ewing’s sarcomas by the work of Grohar et al whose approach deserves further investigation (Grohar and Helman, 2013). On the other hand, the specific pattern of sensitivity of tumour cells deficient in DNA repair mechanisms opens the possibility to new therapeutic strategies. For example, based on the NER profile, it is possible to envisage the sequential administration of trabectedin followed by drugs such as cisplatin; clinical results in ovarian cancer appear to support this strategy (Callata et al, 2013). Also, the high sensitivity of cells that are deficient in HR repair mechanisms suggests that the drug can be particularly effective against breast or ovarian tumours with BRCA1 or BRCA2 mutations (Schoffski et al, 2011; Romano et al, 2013).
Certainly, the ability of trabectedin to decrease TAM and to modulate the production of cytokines and angiogenic factors is also of relevance. Several clinical and preclinical observations advocate a significant effect of trabectedin on the TME (Galmarini et al, 2014). It has also been observed that the response to trabectedin often takes long time, becoming evident only after several courses of therapy, a finding that is difficult to explain whether the drug acts only on cancer cells (Grosso et al, 2007; Sanfilippo et al, 2011; Sanfilippo and Casali, 2013). Changes in tumour density before size reduction have been documented (Hollebecque et al, 2010; Sanfilippo et al, 2011). For instance, in liposarcomas, a decrease in tumour density may occur without tumour shrinkage, eventually resulting in size decrease only after several courses (Grosso et al, 2007; Sanfilippo and Casali, 2013). Furthermore, the drug often induces a very prolonged disease stabilisation indicating that trabectedin appears to keep tumours under control, suggestive of an oncostatic effect (Del Campo et al, 2013; Ferrandina et al, 2013; Le Cesne, 2013). This was documented in a series of studies carried out by the Gynecologic Oncology Group (GOG) group with uterine leiomyosarcoma patients treated with different drugs. Trabectedin showed an increased PFS over any other treatment despite a lower rate of tumour reduction in the same time of study (Monk et al, 2012). This remarkable observation demonstrates a distinct pattern of response for trabectedin when compared with classical agents like ifosfamide, doxorubicin and gemcitabine.
Furthermore, D’Incalci et al (2012) reported significant correlations between patient weight gain and improved survival. Positive correlations occurred during the first cycles of treatment and included small weight differences, suggesting that weight gain is a visible effect of other underlying changes induced by the treatment. The possibility that responders to trabectedin experience changes in inflammatory cytokines such as IL-6, known to be downmodulated by the drug (Allavena et al, 2005), coupled to the fact that this favours weight gain, is worthy of further testing in treated patients.
More studies are needed to elucidate the overall effects of trabectedin on different immunological mechanisms, one example being to assess the relationship between the decrease in the number of immune-suppressive TAM and corresponding effects on adaptive immune response mechanisms. Trabectedin could represent a paradigm to be combined with other therapies directed to elicit anti-tumour cytotoxic lymphocytes. It would be also important to understand whether the different mechanisms of action of trabectedin could be dose and/or treatment schedule dependent. It might be hypothesised that, to obtain a more significant and prolonged anti-inflammatory and anti-angiogenic effect, it would be necessary to use a metronomic administration approach. Work is in progress in our laboratories to evaluate whether this is the case at least at the preclinical level.
Conclusions
As reviewed here, trabectedin not only has direct effects against cancer cells but also has host-modulating properties that appear to be of great importance for its therapeutic effect. Strong preclinical and clinical evidence reveals the ability of this drug to decrease the number of TAMs and to modify the TME and angiogenesis at therapeutically relevant doses. Therefore, it seems plausible to hypothesise that the multiple mechanisms of action may have different roles in different tumours, and thus the determinants of the drug action can be dissimilar in the diverse contexts. It is reasonable to believe that there is a relationship between the effects on cancer cells and the effects on the TME, resulting in a therapeutic synergism. Subsequent studies should address how to exploit the unique mechanistic features of trabectedin to combine it either with immunological or microenvironmental modulators or with cytotoxic agents in a rational manner.
References
Allavena P, Mantovani A (2012) Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol 167: 195–205.
Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, Olimpio CO, Bonardi C, Garbi A, Lissoni A, de Braud F, Jimeno J, D’Incalci M (2005) Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res 65: 2964–2971.
Aune GJ, Takagi K, Sordet O, Guirouilh-Barbat J, Antony S, Bohr VA, Pommier Y (2008) Von Hippel-Lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of RNA polymerase II in response to trabectedin. Clin Cancer Res 14: 6449–6455.
Bissell MJ, Hines WC (2011) Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17: 320–329.
Bueren-Calabuig JA, Giraudon C, Galmarini CM, Egly JM, Gago F (2011) Temperature-induced melting of double-stranded DNA in the absence and presence of covalently bonded antitumour drugs: insight from molecular dynamics simulations. Nucleic Acids Res 39: 8248–8257.
Callata H, Manzano A, Sanchiz B, Alonso T, Gajate P, Sotelo M, Cabezas S, Aguado C, Diaz-Rubio E, Casado A (2013) Treatment with trabectedin enables retreatment with platinum-based chemotherapy in platinum-resistant and refractory (PRR) and partially platinum-sensitive (PPS) recurrent ovarian cancer (ROC) patients. ESGO 18th International Meeting: Liverpool, UK; 144.
Cuevas C, Francesch A (2009) Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat Prod Rep 26: 322–337.
Charytonowicz E, Terry M, Coakley K, Telis L, Remotti F, Cordon-Cardo C, Taub RN, Matushansky I (2012) PPARgamma agonists enhance ET-743-induced adipogenic differentiation in a transgenic mouse model of myxoid round cell liposarcoma. J Clin Invest 122: 886–898.
D’Incalci M, Galmarini CM (2010) A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther 9: 2157–2163.
D’Incalci M, Galmarini CM, de la Riba Alvarez I, Badri N, Schoffski P (2012) Association between body weight and efficacy outcomes during trabectedin therapy for recurrent advanced soft tissue sarcoma (STS). J Clin Oncol 30 (suppl): abstr 10047.
Damia G, D’Incalci M (2007) Targeting DNA repair as a promising approach in cancer therapy. Eur J Cancer 43: 1791–1801.
Damia G, Silvestri S, Carrassa L, Filiberti L, Faircloth GT, Liberi G, Foiani M, D’Incalci M (2001) Unique pattern of ET-743 activity in different cellular systems with defined deficiencies in DNA-repair pathways. Int J Cancer 92: 583–588.
Del Campo JM, Sessa C, Krasner CN, Vermorken JB, Colombo N, Kaye S, Gore M, Zintl P, Gomez J, Parekh T, Park YC, McMeekin S (2013) Trabectedin as single agent in relapsed advanced ovarian cancer: results from a retrospective pooled analysis of three phase II trials. Med Oncol 30: 435.
Di Giandomenico S, Frapolli R, Bello E, Uboldi S, Licandro SA, Marchini S, Beltrame L, Brich S, Mauro V, Tamborini E, Pilotti S, Casali PG, Grosso F, Sanfilippo R, Gronchi A, Mantovani R, Gatta R, Galmarini CM, Sousa-Faro JM, D’Incalci M (2013) Mode of action of trabectedin in myxoid liposarcomas. Oncogene e-pub ahead of print 11 November 2013; doi:10.1038/onc.2013.462.
Erba E, Bergamaschi D, Bassano L, Damia G, Ronzoni S, Faircloth GT, D’Incalci M (2001) Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur J Cancer 37: 97–105.
Ferrandina G, Salutari V, Vincenzi B, Marinaccio M, Naglieri E, Loizzi V, Carpano S, Amadio G, Tonini G, Scambia G, Lorusso D (2013) Trabectedin as single agent in the salvage treatment of heavily treated ovarian cancer patients: a retrospective, multicenter study. Gynecol Oncol 130: 505–510.
Feuerhahn S, Giraudon C, Martinez-Diez M, Bueren-Calabuig JA, Galmarini CM, Gago F, Egly JM (2011) XPF-dependent DNA breaks and RNA polymerase II arrest induced by antitumor DNA interstrand crosslinking-mimetic alkaloids. Chem Biol 18: 988–999.
Forni C, Minuzzo M, Virdis E, Tamborini E, Simone M, Tavecchio M, Erba E, Grosso F, Gronchi A, Aman P, Casali P, D’Incalci M, Pilotti S, Mantovani R (2009) Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol Cancer Ther 8: 449–457.
Friedman D, Hu Z, Kolb EA, Gorfajn B, Scotto KW (2002) Ecteinascidin-743 inhibits activated but not constitutive transcription. Cancer Res 62: 3377–3381.
Gago F, Hurley LH (2002) Devising a structural basis for the potent cytotoxic effects of ecteinascidin 743. In: Small Molecule DNA and RNA Binders: From Synthesis to Nuclear Acid Complexes, Demeunynck M, Bailly C, Wilson WD, (eds) pp 643–675. Wiley-VCH: Weinheim, Germany.
Galmarini CM, D’Incalci M, Allavena P (2014) Trabectedin and plitidepsin: drugs from the sea that strike the tumor microenvironment. Mar Drugs 12: 719–733.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, Nebuloni M, van Rooijen N, Mortarini R, Beltrame L, Marchini S, Fuso Nerini I, Sanfilippo R, Casali PG, Pilotti S, Galmarini CM, Anichini A, Mantovani A, D’Incalci M, Allavena P (2013) Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23: 249–262.
Germano G, Frapolli R, Simone M, Tavecchio M, Erba E, Pesce S, Pasqualini F, Grosso F, Sanfilippo R, Casali PG, Gronchi A, Virdis E, Tarantino E, Pilotti S, Greco A, Nebuloni M, Galmarini CM, Tercero JC, Mantovani A, D’Incalci M, Allavena P (2010) Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res 70: 2235–2244.
Grohar PJ, Griffin LB, Yeung C, Chen QR, Pommier Y, Khanna C, Khan J, Helman LJ (2011) Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 13: 145–153.
Grohar PJ, Helman LJ (2013) Prospects and challenges for the development of new therapies for Ewing sarcoma. Pharmacol Ther 137: 216–224.
Grohar PJ, Segars LE, Yeung C, Pommier Y, D’Incalci M, Mendoza A, Helman LJ (2013) Dual targeting of EWS-FLI1 activity and the associated DNA damage response with Trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growth. Clin Cancer Res 20 (5): 1190–1203.
Grosso F, Jones RL, Demetri GD, Judson IR, Blay JY, Le Cesne A, Sanfilippo R, Casieri P, Collini P, Dileo P, Spreafico C, Stacchiotti S, Tamborini E, Tercero JC, Jimeno J, D’Incalci M, Gronchi A, Fletcher JA, Pilotti S, Casali PG (2007) Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 8: 595–602.
Grosso F, Sanfilippo R, Virdis E, Piovesan C, Collini P, Dileo P, Morosi C, Tercero JC, Jimeno J, D’Incalci M, Gronchi A, Pilotti S, Casali PG (2009) Trabectedin in myxoid liposarcomas (MLS): a long-term analysis of a single-institution series. Ann Oncol 20: 1439–1444.
Herrero AB, Martin-Castellanos C, Marco E, Gago F, Moreno S (2006) Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res 66: 8155–8162.
Hollebecque A, Adenis A, Taieb S, Lebedinsky C, Penel N (2010) Inadequacy of size-based response criteria to assess the efficacy of trabectedin among metastatic sarcoma patients. Invest New Drugs 28: 529–530.
Hurley LH, Zewail-Foote M (2001) The antitumor agent ecteinascidin 743: characterization of its covalent DNA adducts and chemical stability. Adv Exp Med Biol 500: 289–299.
Jin S, Gorfajn B, Faircloth G, Scotto KW (2000) Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc Natl Acad Sci USA 97: 6775–6779.
Le Cesne A (2013) 13 years of trabectedin, 5 years of Yondelis(R): what have we learnt? Expert Rev Anticancer Ther 13: 11–19.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555.
Minuzzo M, Marchini S, Broggini M, Faircloth G, D’Incalci M, Mantovani R (2000) Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc Natl Acad Sci USA 97: 6780–6784.
Monk BJ, Blessing JA, Street DG, Muller CY, Burke JJ, Hensley ML (2012) A phase II evaluation of trabectedin in the treatment of advanced, persistent, or recurrent uterine leiomyosarcoma: a gynecologic oncology group study. Gynecol Oncol 124: 48–52.
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4: 71–78.
Qian BZ, Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141: 39–51.
Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, Garcia-Fernandez LF, Cuevas C, Allavena P, Erba E, D’Incalci M (2013) Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis(R) (PM00104). Int J Cancer 133: 2024–2033.
Sanfilippo R, Casali PG (2013) The intriguing patterns of tumor response to trabectedin. Expert Rev Anticancer Ther 13: 21–24.
Sanfilippo R, Grosso F, Jones RL, Banerjee S, Pilotti S, D’Incalci M, Dei Tos AP, Raspagliesi F, Judson I, Casali PG (2011) Trabectedin in advanced uterine leiomyosarcomas: a retrospective case series analysis from two reference centers. Gynecol Oncol 123: 553–556.
Schoffski P, Taron M, Jimeno J, Grosso F, Sanfilipio R, Casali PG, Cesne AL, Jones RL, Blay JY, Poveda A, Maki RG, Nieto A, Tercero JC, Rosell R (2011) Predictive impact of DNA repair functionality on clinical outcome of advanced sarcoma patients treated with trabectedin: A retrospective multicentric study. Eur J Cancer 47: 1006–1012.
Soares DG, Escargueil AE, Poindessous V, Sarasin A, de Gramont A, Bonatto D, Henriques JA, Larsen AK (2007) Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743. Proc Natl Acad Sci USA 104: 13062–13067.
Takebayashi Y, Pourquier P, Zimonjic DB, Nakayama K, Emmert S, Ueda T, Urasaki Y, Kanzaki A, Akiyama SI, Popescu N, Kraemer KH, Pommier Y (2001) Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat Med 7: 961–966.
Tavecchio M, Simone M, Erba E, Chiolo I, Liberi G, Foiani M, D’Incalci M, Damia G (2008) Role of homologous recombination in trabectedin-induced DNA damage. Eur J Cancer 44: 609–618.
Acknowledgements
Most of the experimental work of MD and PA has been supported by the Italian Association for Cancer Research (AIRC). We would like to thank Maura Montani, Stefania Filippeschi and José Alberto Nadal for their help in the editing and in the reference selection.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
D’Incalci, M., Badri, N., Galmarini, C. et al. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer 111, 646–650 (2014). https://doi.org/10.1038/bjc.2014.149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.149
Keywords
This article is cited by
-
Host obesity alters the ovarian tumor immune microenvironment and impacts response to standard of care chemotherapy
Journal of Experimental & Clinical Cancer Research (2023)
-
DNA repair and damage pathways in mesothelioma development and therapy
Cancer Cell International (2022)
-
Inflammation and tumor progression: signaling pathways and targeted intervention
Signal Transduction and Targeted Therapy (2021)
-
The potential role of the extracellular matrix in the activity of trabectedin in UPS and L-sarcoma: evidences from a patient‐derived primary culture case series in tridimensional and zebrafish models
Journal of Experimental & Clinical Cancer Research (2021)
-
Efficacy and safety of lurbinectedin and doxorubicin in relapsed small cell lung cancer. Results from an expansion cohort of a phase I study
Investigational New Drugs (2021)
