
Abstract
Background:
This trial evaluated the feasibility and efficacy of combined sorafenib and irinotecan (NEXIRI) as second- or later-line treatment of patients with KRAS-mutated metastatic colorectal cancer (mCRC), who had progressed after irinotecan-based chemotherapy.
Methods:
In Phase I, in a 3+3 dose escalation schedule, patients received irinotecan (125, 150 or 180 mg m−2 every 2 weeks), in combination with 400 mg sorafenib b.d. The primary end point was the maximum-tolerated dose of irinotecan. In Phase II, the primary end point was disease control rate (DCR). Secondary end points were progression-free survival (PFS), overall survival (OS) and toxicity.
Results:
Phase I included 10 patients (median age 63 (49–73)); no dose-limiting toxicity was seen. In Phase II, 54 patients (median age 60 (43–80) years) received irinotecan 180 mg m−2 every 2 weeks with sorafenib 400 mg b.d. Nine patients (17%) remained on full-dose sorafenib. The DCR was 64.9% (95% CI, 51–77). Median PFS and OS were 3.7 (95% CI, 3.2–4.7) and 8.0 (95% CI, 4.8–9.7) months, respectively. Toxicities included Grade 3 diarrhoea (37%), neutropenia (18%), hand-foot syndrome (13%) and Grade 4 neutropenia (17%).
Conclusion:
The NEXIRI regimen showed promising activity as second- or later-line treatment in this heavily pretreated mCRC population (ClinicalTrials.gov NCT00989469).
Similar content being viewed by others
Main
Each year, there are an estimated 1.2 million new cases of colorectal cancer worldwide, with the mortality rate reaching 600 000 (Ferlay et al, 2010). Approximately, half of the patients develop metastases during the course of the disease (Van Cutsem et al, 2010), and for the majority of these patients, treatment is mainly palliative. Over the past 10 years, there has been increasing interest in the combination of chemotherapy (e.g., fluoropyrimidines, irinotecan or oxaliplatin) and targeted therapy (e.g., bevacizumab, cetuximab or panitumumab) in the treatment of metastatic colorectal cancer (mCRC) (Cunningham et al, 2004; Hurwitz et al, 2004; Giantonio et al, 2007; Van Cutsem et al, 2009, 2011; Douillard et al, 2010; Peeters et al, 2010). These combinations have become the standard treatment and resulted in significant improvement in response rate (Cunningham et al, 2004; Hurwitz et al, 2004; Giantonio et al, 2007; Van Cutsem et al, 2009, 2011; Peeters et al, 2010), progression-free survival (PFS) (Cunningham et al, 2004; Hurwitz et al, 2004; Giantonio et al, 2007; Van Cutsem et al, 2009, 2011; Douillard et al, 2010; Peeters et al, 2010) and overall survival (OS) (Hurwitz et al, 2004; Giantonio et al, 2007; Van Cutsem et al, 2009, 2011).
In the era of therapeutic personalisation, the identification of molecular biomarkers has a key role in determining both optimal treatment strategies and clinical outcome in patients with mCRC. The presence of the KRAS gene mutation is a well-established predictive factor for lack of response to anti-epidermal growth factor receptor (EGFR) therapies, regardless of the line of treatment (Benvenuti et al, 2007; Di Fiore et al, 2007; Lièvre et al, 2008). At the time of the trial design, there were no effective therapies targeted specifically against KRAS mutant cancers (Tejpar et al, 2012). Indeed, in patients who have disease progression (DP) after irinotecan- or oxaliplatin-based chemotherapies combined with bevacizumab, the addition of cetuximab to irinotecan (Cunningham et al, 2004), or cetuximab or panitumumab as monotherapy are efficacious only in wild-type KRAS tumours (Amado et al, 2008; Karapetis et al, 2008).
Sorafenib is an oral inhibitor of tumour cell proliferation and angiogenesis, and the lead compound in a series of Raf signalling pathway inhibitors. It has already proved its efficacy in refractory kidney cancer and unresectable hepatocellular carcinoma (Llovet et al, 2008; Escudier et al, 2009). Even in the presence of KRAS mutation, sorafenib potently inhibits activation of the mitogen-activated-protein kinase pathway and extracellular signal-regulated phosphorylation by inhibiting the serine threonine kinases Raf-1 and B-Raf. In addition, sorafenib inhibits the receptor tyrosine kinase activity of vascular endothelial growth factors (VEGFR 1, 2 and 3) and platelet-derived growth factor receptor beta (Wilhelm et al, 2004). In preclinical models, sorafenib has demonstrated antitumour activity in colorectal cancer cell lines, including those with KRAS-mutated tumours (Wilhelm et al, 2004; Martinelli et al, 2010). In humans, only a Phase I trial showed the feasibility of combining a weekly schedule of irinotecan 125 mg m−2 (D1,8,15,22 D1=D42) and a fixed dose of sorafenib 400 mg twice daily in 34 patients with solid tumours (23 of whom had colorectal cancer) (Mross et al, 2007). However, preclinical promising results had already been gathered on the synergistic effect of this combination particularly in KRAS-mutated tumours (personal communication at the time) and were recently published (Mazard et al, 2013).
Novel salvage strategies are needed for improving outcomes in selected KRAS-mutated patients who progress after the failure of all approved standard therapies. In addition, combination schedules need to be evaluated in order to help overcome resistance to chemotherapy. On the basis of the above preclinical and clinical data, we conducted a Phase I/II trial with the aim of assessing the feasibility and efficacy of the combined use of sorafenib and the usual 2-week irinotecan regimen as a second- or later-line treatment of patients with mCRC and KRAS-mutated tumours. In addition, we investigated sorafenib’s pharmacokinetic profile, protein expression on tumour tissues and patient genotypes and their association with efficacy and tolerability.
Materials and methods
This was an open-label, single-arm, multicentre Phase I/II trial. The protocol was approved by the local ethics committee (Comité de Protection des Personnes Sud Méditerranée IV, Montpellier, France, EudraCT number 2008-004285-53) and the French competent authority (Agence Nationale de Sécurité du Médicament et des produits de santé). The trial was registered at ClinicalTrials.gov (NCT00989469).
Patients
Eligible patients had histologically confirmed colorectal cancer with measurable unresectable metastatic lesions and documented DP after irinotecan-based chemotherapy and at least one line of chemotherapy. Patients also had centralised confirmation of KRAS mutation status in codons 12 or 13 in the primary tumour or metastases according to Laurent-Puig et al (2009). Other eligibility criteria included a World Health Organization performance status ⩽2, age ⩾18 years, life expectancy >3 months, adequate organ function, absolute neutrophil count ⩾1500 mm3, platelet count ⩾100 000 mm3, haemoglobin >10 g dl−1, total bilirubin ⩽1.5 × institutional upper limit of normal (ULN), AST/ALT ⩽2.5 × ULN (or <5 ULN for patients with metastatic liver involvement), amylase and lipase <1.5 × ULN, and creatinine <1.5 × ULN. Patients were excluded if there was a history of Gilbert’s syndrome, prior surgery or radiotherapy within 4 weeks before entering the trial, bone-only metastases, symptomatic brain metastases or carcinomatous meningitis, a history of epileptic seizures requiring long-term anticonvulsant therapy, long-term use of cytochrome P450 enzyme-inducing agents, and simultaneous use of other systemic anticancer treatments. Patients with uncontrolled hypertension, unstable coronary syndrome, cardiac arrhythmia or active infection were also excluded. Written informed consent was obtained from each patient before trial entry.
Trial design and treatment
Phase I
Patients were to receive oral sorafenib at a fixed dose of 400 mg twice daily (b.d.) in combination with irinotecan delivered intravenously over 90 min every 2 weeks. There were three dose levels for irinotecan: 125, 150 and 180 mg m−2. A standard 3+3 dose escalation design with three to six patients per dose level was used. The Phase I primary end point was the maximum-tolerated dose (MTD) of irinotecan when administered every 2 weeks with 400 mg sorafenib b.d. The MTD was defined as the dose of irinotecan for which at least one dose-limiting toxicity (DLT) occurred for more than 33% of patients during the first four treatment courses. A cycle of therapy was defined as two courses of irinotecan chemotherapy (28 days).
Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. Dose-limiting toxicities were defined as any Grade 4 non-haematological toxicity (except for vomiting in the absence of adequate prophylaxis), any toxicity requiring a cycle delay of more than 15 days, any Grade 4 haematological toxicity lasting for more than 7 days, any Grade 3-4 febrile neutropenia, and any concomitant sepsis with Grade 3-4 neutropenia.
Phase II
Patients were to be treated at the dose of irinotecan established in Phase I with 400 mg of oral sorafenib b.d. Trial treatments were given until the development of unacceptable toxicity, patient refusal to continue or DP, and could be discontinued in case of life-threatening toxic event. Sorafenib dose reductions to 400 mg per day and then every 2 days were permitted for patients experiencing any Grade 3 and 4 haematological and non-haematological toxicities, or any Grade 2 skin toxicity. Subsequent dose re-escalation up to 400 mg b.d. was allowed. In cases of Grade ⩾2 drug-related toxicity, irinotecan perfusion could be delayed for a maximum of 15 days in the absence of full recovery or resolution to Grade ⩽1. The Phase II primary end point was the disease control rate (DCR), defined as the rate of objective response and stable disease. Target lesions were assessed every 8 weeks by independent review of thorax-abdomen-pelvis computed tomography according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. (Therasse et al, 2000). Complete or partial response had to be confirmed on two consecutive assessments 4 weeks apart. The secondary end points were toxicity, PFS and OS. A complete blood cell and platelet count were requested weekly, whereas physical examination and routine laboratory tests were performed before each administration of irinotecan. The last three patients enrolled in the Phase I trial were included in the Phase II trial analysis.
Ancillary Phase II investigations
Pharmacokinetic analysis of sorafenib
Peripheral blood samples were taken on Day 1 (before starting treatment) and at each scheduled visit (days 14, 28, 56, months 4, 6). The blood was collected into a lithium heparin tube, and centrifuged at 2800 g for 10 min to separate the plasma. The plasma was transferred to polypropylene tubes and frozen at −20 °C until assayed. Maximum plasma concentration (Cmax) of sorafenib was evaluated using High-Performance Liquid Chromatography with UV detection. We investigated for any association between sorafenib’s pharmacokinetic parameters and response to treatment or toxicity.
Patient genotyping and protein expressions on the tumour tissues
Patients were genotyped for two genes and two polymorphisms which have previously been suspected of having a role in clinical outcomes in similar populations (Zhang et al, 2006; Liu et al, 2013). Whole blood was collected at enrolment and genomic DNA was extracted from the peripheral lymphocytes using the QIAamp DNA blood maxi kit (Qiagen, Courtaboeuf, France) according to the manufacturer’s recommendations. The single-nucleotide polymorphism G870A (c.723G>A, rs9344) in the CCND1 gene was identified through PCR-high resolution melting (HRM) curve analysis by using previously described cycling conditions and amplification primers (Ho-Pun-Cheung et al, 2007). The short tandem (TA) repeat variation within the TATA box of the UGT1A1 gene (rs8175347) was studied by fragment analysis of fluorescence-labelled PCR products (protocol available on request).
Expression of EGFR, HER2, PTEN, NF-kB and cyclin D1 was evaluated by immunohistochemistry on 3-μm tissue sections of paraffin-embedded specimens as previously performed in previous studies (Chung et al, 2005; Frattini et al, 2007; Scartozzi et al, 2007; Cascinu et al, 2008; Loupakis et al, 2009). The antibody clones, suppliers, antigen-retrieval procedures, dilutions, staining protocols and cut-point scoring are available on request.
We investigated for any association between genotypes or protein expression and response to treatment or toxicity.
Statistical analysis
In the Phase II trial, 45 patients were required to determine the DCR assuming a Simon two-stage minimax design with α=10% and β=5%. To anticipate potential exclusions or losses, nine more patients were included, giving a 20% leeway. A DCR of 20% (p0) or less would indicate that the treatment combination lacked antitumour activity; a DCR of 40% (p1) or more would be considered promising. Patients were evaluable for tolerability and efficacy if they had received at least one and four courses of treatment, respectively. To proceed to second-stage accrual, four or more patients with disease control among the first 21 patients were required. Promising activity was defined as a partial response or a stable disease observed for 14 patients or more on interim analysis at the end of the second stage. Progression-free survival and OS were estimated using the Kaplan–Meier method.
Associations between response to treatment or toxicity and sorafenib’s pharmacokinetic parameters, patient genotypes or protein expression on tumour tissues were investigated by the χ2 test (or the Fisher test, if applicable). A P-value of 0.05 was considered to indicate statistical significance.
Results
Phase I
Ten patients with a median age of 63 years (range, 49–73 years) were enrolled between February and June 2009 from five French centres. All patients had a good performance status (0 to 1). The primary tumour was located in the colon (90%) or rectum (10%). The experimental regimen was administered as a third- (60%) or later-line treatment. Three, four and three patients received irinotecan at 125, 150 and 180 mg m−2, respectively. A median of 6 (range, 1–12) courses of treatment were delivered. No DLT was seen in any patient. Grade 3 skin toxicity included rash (n=2), hand-foot syndrome (n=2), cutaneous abdominal abscess (n=1) and phlyctena (n=1). Of the patients treated with irinotecan 150 mg m−2, three had Grade 3 diarrhoea, one withdrew consent and one had Grade 4 neutropenia lasting less than 7 days; of the patients treated with irinotecan 180 mg m−2, two had Grade 3 diarrhoea. In total, six patients (67%) required sorafenib dose reduction to 400 mg per day after two courses of treatment because of toxicity. Seven (78%) of nine evaluable patients had stable disease after four courses of treatment. The recommended Phase II dose of irinotecan every 2 weeks was therefore defined to be 180 mg m−2 when combined with 400 mg sorafenib b.d.
Phase II
Patients
Fifty-four patients were recruited between June and December 2009 from 10 French centres (Figure 1). Baseline demographics and disease characteristics are listed in Table 1. The majority of patients had liver metastases (72%) and nearly half of the patients presented with lung metastases. Surgical resection of primary tumour and/or distant metastases had been performed in 47 (87%) and 17 (31%) patients, respectively. See Table 2 for details of prior therapy. All patients had previously received palliative fluorouracil- and irinotecan-based chemotherapy with a median of three lines of treatment per patient (maximum eight lines).

Trial flowchart.
Treatment compliance and treatment-related toxicity
The median number of cycles received per patient was four (range, 1–8). Thirteen patients (24%) completed six or more cycles of treatment. Relative dose intensity was 89% for irinotecan and 61% for sorafenib. Nine patients (17%) received sorafenib at the full dose of 400 mg b.d. throughout the treatment schedule. Forty patients (74%) discontinued treatment for the following reasons: DP (n=26), toxicity (n=6), investigator’s decision (n=5) and withdrawal of consent (n=3).
All patients received at least two courses of treatment and were therefore evaluable for tolerance. There was no toxicity-related death, and most adverse events were Grade 1 or 2. Grade 3 toxicities included diarrhoea (37%), asthenia (22%), neutropenia (18%) and hand-foot syndrome (13%). The main Grade 4 treatment-related toxicity was neutropenia, observed in 16.7% of patients (Table 3). The irinotecan dose was adjusted in 19 patients (35%), and a treatment delay was required in 38 patients (70%).
Response and survival
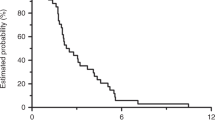
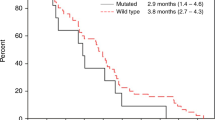
See Table 4 and Figure 2. Tumour response was not evaluable in two patients (3.7%) due to premature treatment discontinuation. The patient who partially responded had previously received 12 cycles of adjuvant chemotherapy combining bevacizumab and FOLFIRI regimen, first-line chemotherapy combining bevacizumab and FOLFOX regimen and second-line treatment with oral capecitabine. The DCR was 64.9% (95% CI, 51–77%) in the intention-to-treat population. Of the nine patients treated with full-dose sorafenib for the whole-treatment schedule, six patients had a stable disease (66.7%). At a median follow-up of 7.0 months (range, 1.2–21.9 months), the median PFS was 3.7 months (95% CI, 3.2–4.7) and the median OS was 8.0 months (95% CI, 4.8–9.7).

Kaplan–Meier curves of (A) progression-free survival and (B) overall survival.
Ancillary Phase II investigations
Sorafenib pharmacokinetics
No relationship was found between treatment response or toxicity and sorafenib’s pharmacokinetic parameters.
Patient genotyping and protein expression on tumour tissues
Among the 48 genomic DNA analysed, 23 displayed the homozygous wild-type UGT1A1 (*1/*1) genotype, 23 the heterozygous UGT1A1 (*1/*28) genotype and two the homozygous polymorphic UGT1A1 (*28/*28) genotype. However, no significant difference in toxicity was observed according to UGT1A1*28 genotype. Analysis of the CCND1 (rs9344) single-nucleotide polymorphism showed that 27.1% of patients were homozygous for the A allele (A/A genotype), 20.8% were homozygous for the G allele (G/G genotype) and 52.1% were heterozygous (A/G genotype). All those variants were distributed according to the Hardy–Weinberg equilibrium. A significant association was found between the CCND1 G870A polymorphism and tumour response in the patients evaluable for efficacy; patients harbouring the A/A variant were significantly associated with stable disease (12/13 patients, P=0.003). We did not detect any association between EGFR, HER2, PTEN, NF-kB or cyclin D1 expression and treatment efficacy or toxicity. CCND1 G870A polymorphisms were not associated with any particular cyclin D1 expression.
Discussion
The results of this Phase I/II trial confirmed the feasibility of combining sorafenib and irinotecan in extensively pretreated patients with mCRC and KRAS mutation. In the Phase I part of the trial, dose escalation of irinotecan was achieved without any DLT, and the recommended Phase II dose was irinotecan 180 mg m−2 once every 2 weeks with fixed-dose sorafenib 400 mg b.d. Principal toxicities included hand-foot syndrome, diarrhoea, asthenia and neutropenia. This treatment combination led to a DCR of 65% (95% CI, 51–77%). Median PFS and OS were 3.7 months and 8.0 months, respectively.
When compared with previously published data in the same kind of heavily pretreated mCRC population with KRAS mutation, our results appear promising. Two Phase III trials assessed the use of the anti-EGFR antibodies cetuximab and panitumumab alone in patients who failed on standard chemotherapy. Both of these studies reported median PFS and OS of 1.9 months and ∼5 months in KRAS-mutated patients, respectively (Amado et al, 2008; Karapetis et al, 2008). More recently, the multitarget drug regorafenib has been evaluated in a randomized Phase III trial as second- or later-line treatment of mCRC with KRAS-mutated or wild-type tumours (Grothey et al, 2013). A total of 760 patients were to receive best supportive care with either oral regorafenib (160 mg daily) or placebo. In this study, 48% of patients had been pretreated by four or more lines for metastatic disease, all had received bevacizumab and 54% had KRAS mutation. The DCR was 41% vs 15% (P<0.001), median PFS was 1.9 vs 1.7 months (HR=0.49; 95% CI, 0.42–0.58, P<0.001) and median OS was 6.4 vs 5 months (HR=0.77; 95% CI, 0.64–0.94, P=0.0052) in the regorafenib arm as compared with placebo, respectively. Subgroup analyses revealed that, among patients treated by regorafenib, KRAS wild-type patients seem to have longer OS than those with KRAS-mutated tumours (HR=0.65; 95% CI, 0.48–0.90 vs HR=0.87; 95% CI, 0.67–1.12; P=0.0038, respectively). However, subsequent analysis demonstrated that there is no statistically significant interaction between OS and KRAS status (Van Cutsem and Grothey, 2013).
Our study is limited by the fact that our Phase I definition of DLT was relatively generous, and that dose reductions in sorafenib had been initially planned for cases of Grade 3 diarrhoea. In a trial such as ours using drug combinations, there is the potential for overlapping toxicities, and some of the toxicities reported in Phase I (such as the Grade 3 diarrhoea) would normally have qualified as DLTs. Possibly due to this limitation, only 17% of Phase II patients received full-dose sorafenib 400 mg b.d. Treatment-related toxicities including Grade 3 diarrhoea (37%) and hand-foot syndrome (13%) and Grade 3-4 neutropenia (35%) were primarily responsible for the high frequency of sorafenib dose reductions to 400 mg. This explains the relative dose intensity of 89% for irinotecan and 61% for sorafenib. For future trials investigating the same drug combination, we therefore recommend that diarrhoea is initially managed by reducing the irinotecan dose, starting at 120 mg m−2 and progressively increasing in case of good tolerance. Compliance to sorafenib 400 mg b.d. could therefore be improved.
Although there are currently no specific biomarkers predicting response to sorafenib, our exploratory studies demonstrated a significant association between the common G870A polymorphism of CCND1 and stabilising of the disease under the therapy. In particular, the A/A genotype was associated with better disease control on univariate analyses, suggesting cyclin D1 as a potential biomarker of the combination.
Our trial supports preclinical data indicating synergistic antitumour effects of combined sorafenib and irinotecan. Sorafenib appears to retain NF-kappa-B in the cytoplasm, and the drug combination may inhibit NF-kappa-B activation, resulting in an enhanced cytotoxicity of irinotecan (Azad et al, 2013). Other studies have shown that multi-tyrosine kinase inhibitors such as sorafenib may also reverse irinotecan resistance (Mross et al, 2007; Azad et al, 2013). Indeed, sorafenib has been identified both in vitro and in vivo as an inhibitor of the drug-efflux pump ABCG2, favouring irinotecan intracellular accumulation thereby enhancing its toxicity (Mazard et al, 2013). This chemosensitizing property previously described by Wei et al (2012) has also been ascribed to the inhibition of the irinotecan-mediated p38 and ERK activation (Mazard et al, 2013).
Possible pharmacokinetic interactions between irinotecan and sorafenib also need to be considered. Our pharmacokinetic investigation was limited to sorafenib, and did not reveal any correlation between treatment-related toxicity and efficacy. However, Mross et al (2007) showed that sorafenib administered at 400 mg b.d. significantly increases irinotecan and SN38 exposure. Another pharmacokinetic study suggested a correlation between sorafenib and exposure to its metabolites with OS and DLTs in 18 patients with CRC, who had been treated with irinotecan at a recommended dose of 100 mg m−2 IV D1, D8 (D1=D42), cetuximab according to the standard weekly schedule, and sorafenib 400 mg b.d. (Azad et al, 2013). Whatever the mechanisms involved, further investigation of this combination appears to be warranted to confirm its efficacy. Pharmacokinetic studies are particularly needed to better characterise any synergy between the two agents. Supported by the preclinical data and this trial’s results, the efficacy of the combination is being further tested in an ongoing multicentre randomized Phase II trial (ClinicalTrials.gov NCT01715441).
Change history
04 March 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26: 1626–1634.
Azad N, Dasari A, Arcaroli J, Taylor GE, Laheru DA, Carducci MA, McManus M, Quackenbush K, Wright JJ, Hidalgo M, Diaz Jr LA, Donehower RC, Zhao M, Rudek MA, Messersmith WA (2013) Phase I pharmacokinetic and pharmacodynamic study of cetuximab, irinotecan and sorafenib in advanced colorectal cancer. Invest New Drugs 31: 345–354.
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A (2007) Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 67: 2643–2648.
Cascinu S, Berardi R, Salvagni S, Beretta GD, Catalano V, Pucci F, Sobrero A, Tagliaferri P, Labianca R, Scartozzi M, Crocicchio F, Mari E, Ardizzoni A (2008) A combination of gefitinib and FOLFOX-4 as first-line treatment in advanced colorectal cancer patients. A GISCAD multicentre phase II study including a biological analysis of EGFR overexpression, amplification and NF-kB activation. Br J Cancer 98: 71–76.
Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, Klimstra DS, Fridman D, Kelsen DP, Saltz LB (2005) Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 23: 1803–1810.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351: 337–345.
Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, Michot F, Michel P, Frebourg T (2007) Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 96: 1166–1169.
Douillard J-Y, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon J-L, Rother M, Oliner KS, Wolf M, Gansert J (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28: 4697–4705.
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Anderson S, Hofilena G, Shan M, Pena C, Lathia C, Bukowski RM (2009) Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 27: 3312–3318.
Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893–2917.
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L (2007) PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 97: 1139–1145.
Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA, Benson 3rd AB (2007) Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 25: 1539–1544.
Grothey A, Cutsem EV, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz H-J, Goldberg RM, Sargent DJ, Cihon F, Cupit L, Wagner A, Laurent D (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381: 303–312.
Ho-Pun-Cheung A, Assenat E, Thezenas S, Bibeau F, Rouanet P, Azria D, Cellier D, Grenier J, Ychou M, Senesse P, Lopez-Crapez E (2007) Cyclin D1 gene G870A polymorphism predicts response to neoadjuvant radiotherapy and prognosis in rectal cancer. Int J Radiat Oncol Biol Phys 68: 1094–1101.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342.
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au H-J, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359: 1757–1765.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet J-B, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouché O, Reid J, Stone S, Penault-Llorca F (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27: 5924–5930.
Lièvre A, Bachet J-B, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, André T, Bibeau F, Diebold M-D, Rougier P, Ducreux M, Tomasic G, Emile J-F, Penault-Llorca F, Laurent-Puig P (2008) KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 26: 374–379.
Liu X, Cheng D, Kuang Q, Liu G, Xu W (2013) Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J e-pub ahead of print 26 March 2013; doi:10.1038/tpj.2013.10.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, de Oliveira AC, Santoro A, Raoul J-L, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz J-F, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359: 378–390.
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, Canestrari E, Funel N, Schiavon G, Petrini I, Magnani M, Tonini G, Campani D, Floriani I, Cascinu S, Falcone A (2009) PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol Off J Am Soc Clin Oncol 27: 2622–2629.
Martinelli E, Troiani T, Morgillo F, Rodolico G, Vitagliano D, Morelli MP, Tuccillo C, Vecchione L, Capasso A, Orditura M, De Vita F, Eckhardt SG, Santoro M, Berrino L, Ciardiello F (2010) Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin Cancer Res Off J Am Assoc Cancer Res 16: 4990–5001.
Mazard T, Causse A, Simony J, Leconet W, Vezzio-Vie N, Torro A, Jarlier M, Evrard A, Del Rio M, Assenat E, Martineau P, Ychou M, Robert B, Gongora C (2013) Sorafenib overcomes irinotecan resistance in colorectal cancer by inhibiting the ABCG2 drug-efflux pump. Mol Cancer Ther 12 (10): 2121–2134.
Mross K, Steinbild S, Baas F, Gmehling D, Radtke M, Voliotis D, Brendel E, Christensen O, Unger C (2007) Results from an in vitro and a clinical/pharmacological phase I study with the combination irinotecan and sorafenib. Eur J Cancer 43: 55–63.
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJA, Strickland AH, Wilson G, Ciuleanu T-E, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28: 4706–4713.
Scartozzi M, Bearzi I, Pierantoni C, Mandolesi A, Loupakis F, Zaniboni A, Catalano V, Quadri A, Zorzi F, Berardi R, Biscotti T, Labianca R, Falcone A, Cascinu S (2007) Nuclear factor-kB tumor expression predicts response and survival in irinotecan-refractory metastatic colorectal cancer treated with cetuximab-irinotecan therapy. J Clin Oncol Off J Am Soc Clin Oncol 25: 3930–3935.
Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E (2012) Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol 30: 3570–3577.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216.
Van Cutsem E, Grothey A (2013) Regorafenib for metastatic colorectal cancer—Authors’ reply. Lancet 381: 1538–1539.
Van Cutsem E, Köhne C-H, Hitre E, Zaluski J, Chang Chien C-R, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360: 1408–1417.
Van Cutsem E, Köhne C-H, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F (2011) Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 29: 2011–2019.
Van Cutsem E, Nordlinger B, Cervantes A (2010) Advanced colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann Oncol 21 (Suppl 5): v93–v97.
Wei Y, Ma Y, Zhao Q, Ren Z, Li Y, Hou T, Peng H (2012) New use for an old drug: inhibiting ABCG2 with sorafenib. Mol Cancer Ther 11: 1693–1702.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64: 7099–7109.
Zhang W, Gordon M, Press OA, Rhodes K, Vallböhmer D, Yang DY, Park D, Fazzone W, Schultheis A, Sherrod AE, Iqbal S, Groshen S, Lenz H-J (2006) Cyclin D1 and epidermal growth factor polymorphisms associated with survival in patients with advanced colorectal cancer treated with Cetuximab. Pharmacogenet Genomics 16: 475–483.
Acknowledgements
We would like to acknowledge the editorial assistance of Vanessa Guillaumon, Catriona Holmes and Julie Courraud. We also thank Patrick Chalbos for his significant work as Clinical Research Associate. This research was supported by a grant from Bayer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Marc Ychou has served on the Bayer board, Olivier Bouché has received honoraria from Bayer and served on the Pfizer board, and Pierre Laurent-Puig has served on the Amgen, Pfizer and Merck boards. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Samalin, E., Bouché, O., Thézenas, S. et al. Sorafenib and irinotecan (NEXIRI) as second- or later-line treatment for patients with metastatic colorectal cancer and KRAS-mutated tumours: a multicentre Phase I/II trial. Br J Cancer 110, 1148–1154 (2014). https://doi.org/10.1038/bjc.2013.813
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.813
Keywords
This article is cited by
-
Advances in the structure, mechanism and targeting of chemoresistance-linked ABC transporters
Nature Reviews Cancer (2023)
-
Phase I dose escalation study of sorafenib plus S-1 for advanced solid tumors
Scientific Reports (2021)
-
Stellenwert der BRAF-Inhibition bei soliden Tumoren wie dem kolorektalen Karzinom
Der Onkologe (2020)
-
Antiangiogenic tyrosine kinase inhibitors in colorectal cancer: is there a path to making them more effective?
Cancer Chemotherapy and Pharmacology (2017)
-
New trends for overcoming ABCG2/BCRP-mediated resistance to cancer therapies
Journal of Experimental & Clinical Cancer Research (2015)
