
Abstract
Background:
Gallbladder cancers and cholangiocarcinomas make up a heterogenous group of tumours with a poor prognosis in advanced stages. On the basis of evidence of dysregulation of the epidermal growth factor receptor, vascular endothelial growth factor and mitogen-activated protein kinase pathways in biliary cancers, we performed a phase 2 trial of sorafenib and erlotinib in patients with advanced biliary cancers.
Methods:
Eligible patients were previously untreated in the advanced setting with adequate hepatic and bone marrow function. Sorafenib and erlotinib were administered continuously at 400 mg BID and 100 mg daily, respectively.
Results:
Thirty-four eligible patients were recruited. The study was terminated after the first stage of accrual owing to failure to meet the predetermined number of patients who were alive and progression free at 4 months. There were two unconfirmed partial responses (6%, 95% CI: 1–20%), with a median progression-free survival of 2 months (95% CI: 2–3), and median overall survival of 6 months (95% CI: 3–8 months). Grade 3 and 4 adverse events included hypertension, AST/ALT increase, bilirubin increase, diarrhoea, hypokalaemia, hypophosphatemia and rash.
Conclusions:
Despite compelling preclinical rationale, the combination of sorafenib and erlotinib does not have promising clinical activity in an unselected population of patients with biliary cancers. Improved patient selection based on tumour biology and molecular markers is critical for future evaluation of targeted therapies in this disease.
Similar content being viewed by others


Main
Biliary tract cancers represent a heterogenous group of neoplasms including intrahepatic and extrahepatic bile duct cancers (cholangiocarcinomas) and gallbladder cancers. The American Cancer Society estimates that 10 310 new cases of gallbladder and bile duct cancers (excluding bile ducts within the liver) will be diagnosed in 2013 in the United States. Similarly, there are predicted to be 30 640 new cases of liver and intrahepatic biliary cancers in 2013, about 15% of which are intrahepatic cholangiocarcinomas (IHCC; Siegel et al, 2013).
Advances in the treatment of unresectable and metastatic biliary cancers have been limited by several factors including the heterogeneity of the disease and incomplete understanding of biliary molecular carcinogenesis. Furthermore, most cytotoxic and targeted drugs had been evaluated for efficacy in the setting of phase 2 studies until the report by Valle et al (2010) established the superiority of gemcitabine and cisplatin over gemcitabine alone, with increases in both progression-free survival (PFS; 8.0 vs 5 months, P<0.001) and overall survival (OS; 11.7 vs 8.1 months, P<0.001) in a large randomised phase 3 trial.
Given the modest benefit of cytotoxic chemotherapy, recent efforts have focused on understanding the molecular carcinogenesis of biliary cancers and the identification of novel therapeutic targets. The epidermal growth factor receptor (EGFR) and vascular endothelial growth factor (VEGF) are both potential targets in biliary cancers. Overexpression of EGFR and VEGF has been reported in 27 and 54% of IHCC samples and in 19 and 59% of extrahepatic cholangiocarcinomas (EHCC). Overexpression of EGFR is associated with poor prognostic features including lymph node metastasis, lymphatic invasion and perineural invasion in EHCC, whereas VEGF overexpression is associated with intrahepatic metastasis in IHCC. The expression of EGFR has also been shown to be a significant independent prognostic factor for recurrence and OS in IHCC (Yoshikawa et al, 2008). In another study, VEGF was found to be expressed in 100% of 19 tumour specimens from patients with cholangiocarcinoma (Benckert et al, 2003).
In addition to the potential role of the VEGF and EGFR pathways in biliary carcinogenesis, the Ras–Raf–Mek–Erk pathway is also thought to modulate biliary tumour growth. For instance, one study showed BRAF mutations in 15 out of 69 (22%) patients with cholangiocarcinoma (Tannapfel et al, 2003). Recently, Andersen et al (2012) reported transcriptome analyses of 104 cholangiocarcinoma specimens and identified a poor prognostic subgroup characterised by KRAS mutations, and activation of EGFR and HER2 signalling pathways.
Both sorafenib and erlotinib have been evaluated as single agents for the treatment of patients with biliary cancers. In a phase 2 study of 42 patients with unresectable or metastatic biliary cancer who were treated with erlotinib as a single agent, Philip et al (2006) reported three partial responses and a stable disease rate of 43%. The median time to progression was 2.6 months and the median OS was 7.5 months. We previously reported the results of a phase 2 study of sorafenib in patients with advanced biliary cancers and noted a median PFS of 3 months and a median OS of 9 months (95% CI: 4–12 months), which was comparable to the survival reported with cytotoxic chemotherapy regimens (El-Khoueiry et al, 2012).
Given these data, we designed and conducted a phase 2 study evaluating the combination of sorafenib and erlotinib based on the modest clinical activity of each agent alone and the extensive molecular cross-talk between the EGFR and VEGF pathways (Ciardiello et al, 2006). A phase 1 study of the combination of sorafenib and erlotinib had been reported by Duran and colleagues (Quintela-Fandino et al, 2010) and determined the maximum tolerated dose to be sorafenib 400 mg PO twice daily and erlotinib 150 mg once daily. At this dose level, six out of seven patients needed a dose delay and five out of seven required a dose reduction at cycle 2 or higher. The toxicities and subsequent dose reductions represented a potential concern for our population, which may be even more susceptible to gastrointestinal toxicities and anorexia. For this reason, we proceeded with sorafenib at 400 mg twice daily and erlotinib at 100 mg daily. Our study was conceived and planned before the initial presentation of the ABC-02 trial results in June of 2009, which established the combination of gemcitabine and cisplatin as a new standard of care for patients with advanced biliary cancers. The study proceeded to activation in April of 2010 as all the stakeholders, including patient advocates, believed that it was important and ethical to explore the efficacy of novel combinations in first-line treatment of biliary cancers as long as they were supported by strong scientific rational. Furthermore, there was agreement that the combination of gemcitabine and cisplatin could be utilised as second-line treatment upon progression that could be determined as early as 8 weeks from the initiation of study therapy.
Materials and methods
Eligibility criteria
Eligible patients had cytologically or pathologically confirmed diagnosis of gallbladder carcinoma or cholangiocarcinoma that was unresectable or metastatic. Prior treatment for metastatic or unresectable disease was not allowed; previous adjuvant therapy was allowed but must have been completed at least 6 months before the documented recurrence. Measurable disease was required. Other eligibility criteria included a Zubrod performance status of 0–1; a measured or calculated creatinine clearance ⩾60 ml min−1; adequate bone marrow function indicated by a leukocyte count ⩾3000 mcl−1, absolute neutrophil count ⩾1000 mcl−1 and platelet count ⩾100 000 mcl−1; adequate hepatic function with a total bilirubin up to 1.5 × the institutional upper limit of normal; serum albumin ⩾2.5 g dl−1; AST and ALT levels ⩽2.5 the upper limit of normal or ⩽5 × upper limit of normal in the presence of liver metastases. In the case of patients with decompression of the biliary tree within 14 days of determining eligibility, stability of the bilirubin had to be confirmed by obtaining a second serum bilirubin level 5–7 days after the first value. Patient with uncontrolled hypertension as evidenced by SBP⩾150 mm Hg or DBP⩾100 mm Hg within 28 days before registration were not allowed.
Treatment plan and toxicity assessment
Sorafenib and erlotinib were supplied by the division of Cancer Treatment and Diagnosis, National Cancer Institute (Bethesda, MD, USA). Patients were treated with sorafenib 400 mg orally twice daily and erlotinib 100 mg orally once daily on a continuous basis. One treatment cycle was of 28 days. Patients were seen and evaluated on a weekly basis during cycle 1, and every 2 weeks in cycle 2 and beyond. Toxicities were graded as per the National Cancer Institute Common Terminology Criteria for adverse events version 4.0. The worst grade of toxicity per patient was recorded in each cycle. Specific dose modification and treatment interruption criteria for sorafenib and erlotinib were applied. If one of the two study drugs was temporarily held due to a specific toxicity that was determined by the treating physician to be exclusively related to that drug, the other drug could be continued per protocol. Treatment with both drugs was held for grade 3 or 4 toxicity, including bilirubin elevation, AST/ALT elevation, diarrhoea, neutropaenia and thrombocytopaenia. In the case of hypertension, sorafenib was held for grade 2 symptomatic or grade 3 hypertension and subsequently restarted with one dose level reduction once diastolic BP was ⩽100 mm Hg and symptoms had resolved. Sorafenib was held in the case of a second or higher recurrence of grade 2 palmar–plantar erythrodysesthesia syndrome, or at the first occurrence of a grade 3 or higher toxicity. Symptomatic management for maculopapular or acneiform rash was instituted at first occurrence, and treatment with both drugs was held for intolerable rash of any grade. Both drugs were dose reduced in the event of a recurrence of an intolerable rash.
Two dose reductions were allowed for each drug; for sorafenib, dose level −1 was 200 mg twice daily, and dose level −2 was 200 mg once daily; for erlotinib, dose level −1 was 75 mg once daily and dose level −2 was 50 mg once daily. Patients requiring treatment interruption for more than 4 weeks or requiring more than two dose reductions were removed from protocol treatment.
Disease assessment
Patient response was assessed every 8 weeks using the Response Evaluation Criteria in Solid Tumors classification 1.1. Measurable disease was defined as at least one lesion for which the longest diameter could be accurately measured as ⩾1 cm using spiral computed tomography or magnetic resonance imaging. Measurable lymph nodes had to have a short-axis measurement of ⩾1.5 cm. All other lesions, including ascites and pleural effusions, were considered non-measurable. Patients who met stable disease criteria at least once after study entry at a minimum interval of 6 weeks were considered to have achieved disease stabilisation. Progression-free survival was calculated from the date of registration to the date of first observation of progressive disease, death due to any cause or symptomatic deterioration. Patients known to be alive and progression free were censored at last date of contact.
Statistical considerations
The primary end point of the trial was PFS in patients with advanced gallbladder cancer or cholangiocarcinoma treated with sorafenib and erlotinib. The secondary end points included response probability, OS and toxicity. We assumed that the combination of sorafenib and erlotinib would be of interest for further study if the true median PFS was 8 months or more, and of no further interest if it were 4 months or less. A two-stage design was planned to evaluate PFS. If after the first 25 patients were accrued, we observed 13 or more patients to be alive and without progression at 4 months, the study would accrue an additional 25 patients to the second stage.
Results
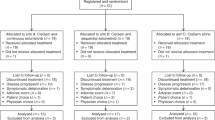
A total of 40 patients were registered between April of 2010 and March of 2011. Six were ineligible because of elevated blood pressure or baseline laboratory abnormalities. The median age of eligible patients was 63 (range 49–82). Thirteen patients (38%) were male and 28 (82%) were white. Fifty-six percent of patients had a Zubrod performance status of 0. Twenty patients had cholangiocarcinoma (59%) and 14 (41%) had gallbladder cancer. Only one patient had received prior adjuvant chemotherapy and no patients had received radiation therapy. Twenty-eight (82%) patients had metastatic disease and six (18%) had locally advanced disease (Table 1).
Administration of sorafenib and erlotinib
The median number of cycles administered was 2 (range 1–14). Reasons for treatment discontinuation included adverse events in 7 patients (21%), progression in 24 (71%) and death in 2 (6%). Dose reduction or interruption secondary to adverse events occurred in 17 out of 34 patients (50%) in cycle 1 and 14 out of 26 patients (54%) in cycle 2. The median dose of sorafenib and erlotinib delivered in cycle 1 was 77% and 98% of the planned dose, respectively. When combining cycles 1 and 2, the median dose of sorafenib and erlotinib delivered over the first two cycles was 52% and 75%, respectively. It is important to note that the dose intensity was not only affected by dose reduction and interruption for adverse events but also by discontinuation due to disease progression or patient refusal in 8 out of 34 patients (24%).
Toxicity
Thirty-four patients were evaluated for adverse events. One patient died on protocol therapy after being admitted for abdominal pain. The most common grade 3 and 4 toxicities that were at least possibly related to study drugs and occurred in two or more patients were hypertension (15%), ALT increase (12%), alkaline phosphatase increase (9%), diarrhoea (9%), hypophosphatemia (9%), AST increase (9%), bilirubin increase (6%), hand-foot skin reaction (6%), hepatic infection (6%), hypokalaemia (6%) and rash (6%; Table 2).
Efficacy
The study was terminated after the first stage of accrual because of failure to meet the requirement of 13 patients being alive and without progression at 4 months. Thirty-four patients were evaluable for response. Two patients (6%) achieved an unconfirmed partial response. Ten (29%) patients had stable disease. Eighteen (53%) patients had progressive disease, one patient had symptomatic deterioration leading to treatment discontinuation and three patients did not have adequate assessment to determine response. Median PFS was 2 months (95% CI: 2–3 months) and 4 month PFS was 29% (95% CI: 13–45%; Figure 1). Median OS was 6 months (95% CI: 3–8 months; Figure 2). Information about subsequent therapy was available for 32 patients; 13 patients (41%) received systemic treatment after progression on study. The second-line treatments were gemcitabine and a platinum combination in eight patients, gemcitabine and capecitabine combination in two patients, erlotinib in one patient and docetaxel in one patient. Only two patients received third-line treatment.

Progression-free survival.

Overall survival.
Discussion
In this phase 2 trial, we explored the efficacy of the combination of erlotinib and sorafenib in patients with unresectable or metastatic gallbladder cancer and cholangiocarcinoma. The study was closed after the first stage of accrual owing to failure to meet predetermined criteria for efficacy. The median PFS and OS noted with sorafenib and erlotinib were not superior to those noted in phase 2 studies that evaluated erlotinib and sorafenib as single agents in patients with biliary cancers. Our study was based on the hypothesis that simultaneous inhibition of the VEGF, EGFR and mitogen-activated protein kinase pathways would result in improved outcomes for patients with biliary cancers. Preclinical data provide evidence of extensive interaction between the VEGF and EGFR pathways and support the hypothesis that oncogenic properties of the EGFR pathways are, at least in part, driven by proangiogenic factors. Activation of EGFR has been shown to result in upregulation of VEGF-A production in cancer cells (Goldman et al, 1993; Gille et al, 1997). Similarly, secretion of proangiogenic factors including VEGF, interleukin 8 and transforming growth factor-β is inhibited by EGFR blockade (Bruns et al, 2000; Hirata et al, 2002). Moreover, several studies have shown the role of VEGF-A upregulation in the acquired resistance to EGFR treatment in initially EGFR inhibitor-sensitive cancer cells (Viloria-Petit et al, 2001; Ciardiello et al, 2004). Preclinical studies in non-small cell lung cancer cell lines and colon cancer cell lines have revealed synergistic inhibition of colony formation when sorafenib and erlotinib or cetuximab were combined (Martinelli et al, 2010). Despite the robust preclinical rational, our study failed to reveal significant activity for the combination of sorafenib and erlotinib in biliary cancers. This is consistent with results of other clinical trials that evaluated the combination of sorafenib and erlotinib in patients with non-small cell lung cancer and hepatocellular carcinoma (Spigel et al, 2011; Zhu et al, 2012). The median OS of 6 months in our study may represent a detrimental effect of this combination in an unselected patient population, but this remains speculative in the setting of a small single-arm phase 2 study and in the absence of a detailed mechanistic evaluation of the impact of the simultaneous administration of sorafenib and erlotinib. It is also noteworthy that only 40% of patients received second-line treatment in this study. Thus, the evaluation of novel combinations of targeted therapies in first-line treatment of biliary cancers has to be based on robust scientific evidence and with the awareness that not all patients may be able to receive second-line treatment with the standard combination of gemcitabine and cisplatin.
Our study has several limitations, including the relatively small number of patients, the rate of dose interruptions and reductions in cycles 1 and 2, and the compromised dose intensity that was mostly due to adverse events. This observation raises concerns about the tolerability of the combination of sorafenib and erlotinib in patients with advanced biliary cancers. Another limitation of our trial is the heterogeneity of the patient population as a result of the inclusion of all types of biliary cancers, independent of the site of origin. This is of special concern when using targeted therapeutic drugs as the expression of genes involved in cell cycle control, apoptosis and of EGFR and HER2 vary significantly depending on the site of origin of biliary cancers (Jarnagin et al, 2006; Yoshikawa et al, 2008). Therefore, stratification based on tumour location along the biliary tree should be considered for future studies of molecularly targeted therapies. Furthermore, the likelihood of targeted therapies resulting in improved therapeutic outcomes may be optimised by selecting patients whose tumours harbour specific genetic alterations. For instance, in the randomised phase 2 study of sorafenib and erlotinib vs erlotinib in patients with NSCLC, which failed to show improved PFS and OS with the combination, a subset of patients with wild-type (WT) EGFR, had a significant improvement in PFS and OS. The authors hypothesised that EGFR WT tumours are more dependent on other signalling pathways, including VEGFR, Raf or platelet-derived growth factor receptor, which are inhibited by sorafenib (Spigel et al, 2011). As a consequence, the availability and procurement of adequate tissue samples should be strongly considered as part of the eligibility criteria of trials evaluating novel therapies in biliary carcinoma.
Despite the emergence of data about genetic alterations in biliary cancers, several challenges continue to face targeted drug development in this disease. These include the molecular heterogeneity of biliary cancers that may be related to site of origin (intrahepatic vs extrahepatic vs gallbladder carcinoma; Jarnagin et al, 2006) or to the fact that most studies that have examined the frequency of genetic alterations such as KRAS or BRAF have been plagued by small sample sizes and yielded inconsistent results (Kipp et al, 2010; Robertson et al, 2013; Voss et al, 2013). Nonetheless, there are emerging and promising therapeutic targets in cholangiocarcinoma that are under active investigation. For example, two small studies using MEK inhibitors have shown single-agent objective responses and promising survival (Bekaii-Saab et al, 2011; Finn et al, 2012). On the basis of this, a clinical trial evaluating the single-agent MEK inhibitor trametinib in comparison with 5-fluorouracil or capecitabine in patients with biliary cancers after progression on gemcitabine and cisplatin is planned by the Southwest Oncology Group. A comprehensive translational medicine plan is included in this trial with the aim of identifying predictive markers of activity.
Similarly, a randomised study comparing gemcitabine and cisplatin vs gemcitabine and cisplatin in combination with selumitinib is planned in the United Kingdom. This is an important approach as it is currently unknown whether single-agent targeted therapies will achieve sufficient therapeutic benefit in biliary cancers in the absence of an established ‘driver’ target. Another promising target in biliary cancers is the MET oncogene the expression of which has been shown to be an independent predictor of poor survival in patients with cholangiocarcinoma (Miyamoto et al, 2011; Andersen et al, 2012). Evaluation of MET targeting agents in cholangiocarcinoma would be warranted, especially given the promising activity of MET inhibitors in hepatocellular carcinoma where MET expression appears to be associated with the likelihood of benefit (Santoro et al, 2013).
In conclusion, the combination of sorafenib and erlotinib does not have promising clinical activity in an unselected population of patients with biliary cancers. Improved patient selection based on tumour location, tumour biology and molecular markers will be critical for future evaluation of targeted therapies in this heterogenous disease.
Change history
18 February 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, Conner EA, Gillen MC, Roskams T, Roberts LR, Factor VM, Thorgeirsson SS (2012) Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 142: 1021–1031,, e15.
Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, O'Neil BH, Balsom S, Balint C, Liersemann R, Vasko VV, Bloomston M, Marsh W, Doyle LA, Ellison G, Grever M, Ringel MD, Villalona-Calero MA (2011) Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol 29: 2357–2363.
Benckert C, Jonas S, Cramer T, Von Marschall Z, Schafer G, Peters M, Wagner K, Radke C, Wiedenmann B, Neuhaus P, Hocker M, Rosewicz S (2003) Transforming growth factor beta 1 stimulates vascular endothelial growth factor gene transcription in human cholangiocellular carcinoma cells. Cancer Res 63: 1083–1092.
Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, Abbruzzese J, Traxler P, Buchdunger E, Radinsky R, Fidler IJ (2000) Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res 60: 2926–2935.
Ciardiello F, Bianco R, Caputo R, Damiano V, Troiani T, Melisi D, De Vita F, De Placido S, Bianco AR, Tortora G (2004) Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res 10: 784–793.
Ciardiello F, Troiani T, Bianco R, Orditura M, Morgillo F, Martinelli E, Morelli MP, Cascone T, Tortora G (2006) Interaction between the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor (VEGF) pathways: a rational approach for multi-target anticancer therapy. Ann Oncol 17 (Suppl 7): vii109–vii114.
El-Khoueiry AB, Rankin CJ, Ben-Josef E, Lenz HJ, Gold PJ, Hamilton RD, Govindarajan R, Eng C, Blanke CD (2012) SWOG 0514: a phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest New Drugs 30: 1646–1651.
Finn RS, Javle MM, Tan BR, Weekes CD, Bendell JC, Patnaik A, Khan GN, Laheru D, Anderson L, Christy-Bittel JL, Barrett E, Guthrie K, Litwiler KS, Bekaii-Saab TS (2012) A phase I study of MEK inhibitor MEK162 (ARRY-438162) in patients with biliary tract cancer. J Clin Oncol 30 (suppl 4): ): abstr 220.
Gille J, Swerlick RA, Caughman SW (1997) Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO J 16: 750–759.
Goldman CK, Kim J, Wong WL, King V, Brock T, Gillespie GY (1993) Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiforme pathophysiology. Mol Biol Cell 4: 121–133.
Hirata A, Ogawa S, Kometani T, Kuwano T, Naito S, Kuwano M, Ono M (2002) ZD1839 (Iressa) induces antiangiogenic effects through inhibition of growth factor receptor tyrosine kinase. Cancer Res 62: 2554–2560.
Jarnagin WR, Klimstra DS, Hezel M, Gonen M, Fong Y, Roggin K, Cymes K, DeMatteo RP, D'Angelica M, Blumgart LH, Singh B (2006) Differential cell cycle-regulatory protein expression in biliary tract adenocarcinoma: correlation with anatomic site, pathologic variables, and clinical outcome. J Clin Oncol 24: 1152–1160.
Kipp BR, Fritcher EG, Clayton AC, Gores GJ, Roberts LR, Zhang J, Levy MJ, Halling KC (2010) Comparison of KRAS mutation analysis and FISH for detecting pancreatobiliary tract cancer in cytology specimens collected during endoscopic retrograde cholangiopancreatography. J Mol Diagn 12: 780–786.
Martinelli E, Troiani T, Morgillo F, Rodolico G, Vitagliano D, Morelli MP, Tuccillo C, Vecchione L, Capasso A, Orditura M, De Vita F, Eckhardt SG, Santoro M, Berrino L, Ciardiello F (2010) Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin Cancer Res 16: 4990–5001.
Miyamoto M, Ojima H, Iwasaki M, Shimizu H, Kokubu A, Hiraoka N, Kosuge T, Yoshikawa D, Kono T, Furukawa H, Shibata T (2011) Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer 105: 131–138.
Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, Donehower RC, Fitch T, Picus J, Erlichman C (2006) Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol 24: 3069–3074.
Quintela-Fandino M, Le Tourneau C, Duran I, Chen EX, Wang L, Tsao M, Bandarchi-Chamkhaleh B, Pham NA, Do T, MacLean M, Nayyar R, Tusche MW, Metser U, Wright JJ, Mak TW, Siu LL (2010) Phase I combination of sorafenib and erlotinib therapy in solid tumors: safety, pharmacokinetic, and pharmacodynamic evaluation from an expansion cohort. Mol Cancer Ther 9: 751–760.
Robertson S, Hyder O, Dodson R, Nayar SK, Poling J, Beierl K, Eshleman JR, Lin MT, Pawlik TM, Anders RA (2013) The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum Pathol 44 (12): 2768–2773.
Santoro A, Rimassa L, Borbath I, Daniele B, Salvagni S, Van Laethem JL, Van Vlierberghe H, Trojan J, Kolligs FT, Weiss A, Miles S, Gasbarrini A, Lencioni M, Cicalese L, Sherman M, Gridelli C, Buggisch P, Gerken G, Schmid RM, Boni C, Personeni N, Hassoun Z, Abbadessa G, Schwartz B, Von Roemeling R, Lamar ME, Chen Y, Porta C (2013) Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol 14: 55–63.
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA: Cancer J Clin 63: 11–30.
Spigel DR, Burris HA 3rd, Greco FA, Shipley DL, Friedman EK, Waterhouse DM, Whorf RC, Mitchell RB, Daniel DB, Zangmeister J, Bass JD, Hainsworth JD (2011) Randomized, double-blind, placebo-controlled, phase II trial of sorafenib and erlotinib or erlotinib alone in previously treated advanced non-small-cell lung cancer. J Clin Oncol 29: 2582–2589.
Tannapfel A, Sommerer F, Benicke M, Katalinic A, Uhlmann D, Witzigmann H, Hauss J, Wittekind C (2003) Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 52: 706–712.
Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J (2010) Cisplatin plus gemcitabine vs gemcitabine for biliary tract cancer. N Engl J Med 362: 1273–1281.
Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, Rak J, Kerbel RS (2001) Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res 61: 5090–5101.
Voss JS, Holtegaard LM, Kerr SE, Fritcher EG, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR (2013) Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol 44: 1216–1222.
Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, Hirohashi S, Shibata T (2008) Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer 98: 418–425.
Zhu A, Rosmorduc O, Evans J, Ross P, Santoro A, Carrilho FJ, Leberre MA, Jensen M, Meinhardt G, Kang YK (2012) SEARCH: A phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with hepatocellular carcinoma (HCC). LBA 917. Presented at 37th European Society of Medical Oncology annual meeting, 2012, Vienna, Austria.
Acknowledgements
This investigation was supported in part by the following PHS Cooperative Agreement grant numbers awarded by the National Cancer Institute, DHHS: CA32102, CA38926, CA46441, CA46282, CA37981, CA58882, CA58723, CA45807, CA35176, CA35090, CA63848, CA67575, CA20319, CA16385, CA35431, CA13612 and CA63844.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
El-Khoueiry, A., Rankin, C., Siegel, A. et al. S0941: a phase 2 SWOG study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br J Cancer 110, 882–887 (2014). https://doi.org/10.1038/bjc.2013.801
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.801
Keywords
This article is cited by
-
Plasmalemma vesicle-associated protein promotes angiogenesis in cholangiocarcinoma via the DKK1/CKAP4/PI3K signaling pathway
Oncogene (2021)
-
Gallbladder cancer with EGFR mutation and its response to GemOx with erlotinib: a case report and review of literature
World Journal of Surgical Oncology (2020)
-
Cholangiocarcinoma — evolving concepts and therapeutic strategies
Nature Reviews Clinical Oncology (2018)
-
Surgical Management of Hilar Cholangiocarcinoma
Current Surgery Reports (2018)
-
Update on the Diagnosis and Treatment of Cholangiocarcinoma
Current Gastroenterology Reports (2017)
