
Abstract
Background:
The mammalian target of rapamycin (mTOR) pathway is dysregulated in small-cell lung cancer (SCLC) and everolimus is an oral mTOR inhibitor.
Methods:
This phase-1b study assessed everolimus safety at the levels of 2.5, 5, or 10 mg once daily in combination with paclitaxel (175 mg m−2) once every 3 weeks in previously treated SCLC patients. The primary end point was to determine the maximum tolerated dose of everolimus.
Results:
Among 21 enrolled patients, common drug-related adverse events were anaemia, neutropenia, thrombocytopenia, pain, hyperglycemia, and stomatitis. Out of 11 evaluable patients treated with everolimus at the level of 5 mg, 1 patient experienced dose-limiting toxicity (DLT) of grade 4 febrile neutropenia and grade 3 thrombocytopenia. The other two DLTs (grade 4 thrombocytopenia and grade 3 hyperglycemia) occurred in two out of three patients receiving everolimus 10 mg. The overall objective response rate was 28%.
Conclusion:
Everolimus showed an acceptable safety profile and preliminary antitumour activity at the dose of 5 mg once daily when combined with 3-weekly paclitaxel 175 mg m−2 in patients with SCLC.
Similar content being viewed by others

Main
Small-cell lung cancer (SCLC) accounts for approximately 13% of all lung cancer cases (Govindan et al, 2006). Although some chemotherapeutic agents, such as etoposide or topoisomerase inhibitor 1 (irinotecan or topotecan) with or without a platinum compound, achieve initially high response rates (Noda et al, 2002; Hanna et al, 2006), most cases of SCLC progress, leading to high mortality rates; thus, new therapeutic strategies are urgently needed.
The mammalian target of rapamycin (mTOR) functions downstream of the phosphatidylinositol-3 kinase (PI3K)/Akt pathway and is activated in response to the activation of PI3K/Akt signaling (Bjornsti and Houghton, 2004). The PI3K/Akt/mTOR pathway has a central role in regulating cell growth, proliferation, and survival (Sun et al, 2006). Furthermore, it is well documented that this pathway is upregulated in SCLC (Marinov et al, 2009; Schmid et al, 2010).
Everolimus, an orally administered rapamycin analog, is a specific inhibitor of mTOR and is approved for treatment of renal cell cancer, pancreatic neuroendocrine cancer, and breast cancer (Motzer et al, 2008; Yao et al, 2011; Baselga et al, 2012). Everolimus has been shown to inhibit activation of the mTOR pathway and tumour growth in SCLC cell lines and in a xenograft model (Marinov et al, 2009). However, in a recently published phase-2 study, everolimus had limited single-agent antitumour activity in patients with SCLC (Tarhini et al, 2010). Meanwhile, there have been several studies showing improved antitumoural or clinical effects of everolimus when combined with cytotoxic or other targeted agents in various tumour types (Mondesire et al, 2004; Faried et al, 2006; Campone et al, 2009; Marinov et al, 2009; Andre et al, 2010; Ramalingam et al, 2010; Schmid et al, 2010; Morrow et al, 2011; O’Reilly et al, 2011; Xu et al, 2011; Dong et al, 2012; Ren et al, 2012). Therefore, it could be possible for everolimus to be effective when used in a combination regimen for SCLC patients to overcome tumour cell drug resistance.
Paclitaxel remains an important salvage chemotherapy for SCLC. The 3-weekly (once every 3 weeks) paclitaxel regimen resulted in considerable response in patients with refractory SCLC (Smit et al, 1998). However, more than half of the patients succumbed to disease progression within 2 months after starting paclitaxel chemotherapy. Although the resistance mechanism to paclitaxel appears to be multifactorial, there is consistent evidence that the PI3K/Akt/mTOR pathway is associated with resistance to paclitaxel (VanderWeele et al, 2004; Faried et al, 2006). It was shown that the sustained exposure of cancer cells to paclitaxel induced the activation of Akt and its downstream molecule, mTOR, leading to the development of resistance to paclitaxel. Furthermore, this resistance was overcome by the inhibition of mTOR by rapamycin (VanderWeele et al, 2004). Many preclinical studies have produced results supporting the existence of synergism between paclitaxel and mTOR inhibitor (Hu et al, 2002; Mondesire et al, 2004; Faried et al, 2006; O’Reilly et al, 2011).
On the basis of these observations, we hypothesised that administration of everolimus with paclitaxel would improve the clinical outcome in patients with SCLC. Therefore, this study was performed to assess the safety and effectiveness of the combination regimen of everolimus plus paclitaxel in cases of previously treated SCLC.
Materials and Methods
Study design
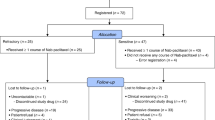
The primary objective of this study was to determine the maximum-tolerated dose (MTD) of everolimus in combination with a fixed dose (175 mg m−2 every 3 weeks) of paclitaxel, based on the incidence of dose-limiting toxicities (DLTs) during the first treatment cycle. The secondary objectives were to characterise the pharmacokinetic disposition of everolimus at the level of the MTD when given in combination with paclitaxel, to evaluate phospho-Akt and phospho-S6 expression as predictive biomarkers for response to everolimus, and to get preliminary information regarding the efficacy of the combination regimen in patients with SCLC. This open-label, single-arm phase-1b study (clinicalTrials.gov ID: NCT01079481) was conducted in accordance with the Declaration of Helsinki and good clinical practice. The study protocol and consent forms were approved by the institutional review board of Samsung Medical Center, and written informed consent was obtained from all participants.
Study population
Eligible patients were adults at least 18 years of age with pathologically confirmed limited or extensive staged SCLC, whose cancer had progressed after at least two prior chemotherapy regimens including at least one platinum-containing regimen. Other eligibility criteria were an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–2, at least one measurable lesion by Response Evaluation Criteria in Solid Tumours (RECIST 1.1), which had not been radiated previously, absolute neutrophil count ⩾1500 per μl, platelet count ⩾1 00 000 per μl, serum total bilirubin ⩽1.5 × institutional upper limit of normal, serum transaminases ⩽5 × upper limit of normal, and creatinine clearance ⩾50 ml min−1. Patients must have terminated prior chemotherapy or radiotherapy 4 weeks before enrolment, and had to have recovered from toxicities related to prior therapies to grade 0–1.
Patients were ineligible if they had received prior therapy with known mTOR inhibitors, had diseases that might alter the absorption of everolimus, or had untreated or symptomatic brain metastases. Patients were also excluded if they had hypercholesterolemia of grade 3 or higher, uncontrolled diabetes with fasting glucose level >165 mg dl−1, uncontrolled infection, liver cirrhosis, or active viral hepatitis. Women who were pregnant, breast-feeding, or able to conceive but unwilling to practice effective contraception and patients with a known Cremophor allergy were also excluded.
Treatment plan
Paclitaxel (Bristol-Myers Squibb Pharmaceuticals Ltd, New York, NY, USA) was administered as a 3-h intravenous infusion every 3 weeks. Everolimus (Novartis Pharmaceuticals, Basel, Switzerland) was given orally in a once-daily dose based on a 21-day treatment course. Patients receiving at least one dose of study drug medications were evaluated weekly for safety during the first cycle of treatment, and they were followed up every 3 weeks. Treatment was continued until disease progression, unacceptable toxicity, or withdrawal of informed consent. After six cycles of combination therapy, patients without disease progression were continued on single-agent everolimus at the same dose and schedule as when given with paclitaxel.
Prophylaxis of emesis and allergic reactions to paclitaxel using serotonin antagonist or steroid was allowed. Prophylactic colony-stimulating factors were not allowed for cycle 1 of therapy.
Dose escalation and adjustment
The safety of the drug combination was explored with the 3-weekly dose of paclitaxel 175 mg m−2 combined initially with 2.5 mg of everolimus daily (dose level 1) and then escalated to 5 mg daily (dose level 2) and 10 mg daily (dose level 3). Up to six patients were enrolled for each individual level. Intrapatient dose escalation was not permitted. Dose escalation decisions were made after every DLT occurred within the first cycle of treatment. If none or one out of six patients experienced a DLT, dose escalation was continued. If two patients experienced DLTs out of the maximum six patients at each level, the toxicity was considered unacceptable and further enrolment was stopped at that level. After the highest well-tolerated dose of everolimus in combination with paclitaxel was established, six more patients were evaluated at the expansion cohort to obtain additional safety and pharmacokinetic information at the level. If three or fewer DLTs occurred out of these 12 patients, this level would be determined as the MTD and the recommended dose for the further phase-2 trials.
For patients who experienced severe toxicities or DLTs, dose modifications were done for the subsequent cycle: the dose of everolimus was reduced to none from 2.5 mg d−1, to 2.5 mg d−1 from 5 mg d−1, or to 5 mg d−1 from 10 mg d−1, and the dose of paclitaxel was reduced by 25% decrements.
Study assessments and DLT
All toxicities were graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. A DLT was defined as a drug-related CTCAE of grade 3 or 4 non-haematological toxicity despite the fully preventive therapy. Additional DLTs included: fever with grade 3 or 4 neutropenia; grade 4 neutropenia lasting for 7 days; grade 3 or 4 thrombocytopenia; neutropenia or thrombocytopenia failing to revert to grade ⩽1 within 3 weeks. Tumour response was assessed every two cycles according to RECIST version 1.1.
Pharmacokinetic measurements
To evaluate the pharmacokinetic parameters of everolimus at the MTD level administered with paclitaxel, blood samples were collected just before the administration of everolimus on days 1, 8, and 15 of cycle 1, and day 1 of cycle 2, and 1, 2, 4, 6, 12, and 24 h after dosing on day 1 of cycle 2 (a total of 10 times per patient) from six patients enrolled in the expansion cohort. Everolimus concentrations were measured in whole blood by liquid chromatography-tandem mass spectrometry after protein precipitation.
Immunohistochemistry
To evaluate PI3K/Akt pathway activation, phospho-Akt and phospho-S6 expressions were assessed by immunohistochemistry. Tumour specimens obtained before treatment initiation were used.
Immunohistochemical studies were carried out on formalin-fixed, paraffin-embedded, 4 μm-thick tissue sections. Tissue sections were deparaffinised three times in xylene for a total of 15 min and subsequently rehydrated. Immunostaining was performed using rabbit monoclonal Phospho-Akt (Ser473)(D9E) XP antibody (#4060, Cell Signaling Technology, Danvers, MA, USA) and phospho-S6 ribosomal protein (Ser235/236) antibody (#2211, Cell Signaling Technology) with the dilution of 1 : 80 and 1 : 200, respectively, and Bond-max autoimmunostainer (Leica Biosystems, Melbourne, Victoria, Australia) with BondTM Polymer refine detection, DS9800 (Vision Biosystems). Staining for phospho-Akt and phospho-S6 was considered to be positive when more than 10% of tumour cells showed membranous reactivity.
Results
Patient population
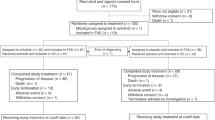
A total of 21 patients with SCLC were enrolled between January 2010 and May 2012. Baseline characteristics were noted at the time of enrollment and are shown in Table 1. The median age of patients enrolled was 59 years. Fourteen patients had an ECOG PS of 1 and seven had a PS of 2. All patients had been previously treated with at least two chemotherapeutic regimens with a median of 11 cycles (range, 7–18). Six patients received prior thoracic radiotherapy, and 11 patients received cranial irradiation (6 for prophylactic purposes and 5 for the control of brain metastasis). The disease was found to relapse after a durable remission of more than 3 months following the completion of first-line chemotherapy in nine patients (sensitive relapse), and the other 12 patients experienced progression within 3 months after completion of first-line chemotherapy (refractory relapse). Ten patients had one metastatic organ, seven had two, and four had three or more metastatic organs.
Toxicity
None of the six patients entered at dose level 1 experienced DLT in cycle 1, allowing escalation to dose level 2. At dose level 2, one out of the six patients experienced DLT (fever with grade 4 neutropenia and grade 3 thrombocytopenia, leading to death) in cycle 1, allowing escalation to dose level 3. At dose level 3, the first (grade 4 thrombocytopenia) and the third (grade 3 hyperglycemia) patients experienced DLTs during cycle 1; thus, level 2 was regarded as the highest well-tolerated dose and this cohort was expanded by an additional six patients to obtain additional safety and pharmacokinetic data. However, one out of the additional six patients experienced tumour progression on day 8 of cycle 1 and was not fully evaluable. As a result, among the 11 patients evaluable for toxicity at level 2, DLT occurred in only 1 patient, as described previously, and level 2 was determined as the MTD and the recommended phase-2 dose.
The median duration of the regimen at dose level 2 was three cycles (range, 1–9): three patients completed six cycles, including two patients who continued to receive single-agent everolimus for 3 and 9 weeks after paclitaxel discontinuation.
Overall, the study treatment was well tolerated at every level and during the whole study period. As described early, there was one drug-related death event due to pneumonia with grade 4 neutropenia, which was counted as a DLT at dose level 2. All grades of toxicities are reported in Table 2.
Antitumour activity and its association with phospho-Akt and phospho-S6 expression
Of the patients enrolled in the study, 18 were evaluable for tumour response. The overall response rate was 28% (5/18) and the amount of tumour shrinkage is shown in Figure 1. All five objective responses were seen in patients who had been categorised as being in the group of sensitive relapse. Although the clinical relevance is limited because of the small sample size, there was a tendency of higher response rates according to the increased everolimus dose level: one objective response out of 6 patients at dose level 1 (17%), 2 out of 10 patients at dose level 2 (20%), and 2 out of 3 patients in dose level 3 (67%).

Maximum change from baseline in tumour burden in the evaluable population ( n =18): the mark (*) indicates the sensitive relapse group regarding the first-line chemotherapy.
Among the six tumour specimens available for phospho-Akt and phospho-S6 staining, two were positive for phospho-Akt and three were positive for phospho-S6, whereas one was positive for both (Figure 2). No objective response was observed in these two phospho-Akt-positive patients (one stable and one progressive disease). On the other hand, two objective responses were observed among the four patients with phospho-Akt-negative tumours. Regarding phospho-S6, one response was, respectively, shown in the phospho-S6-positive (1/3) and -negative (1/3) groups.

Positive immunostaining for both phospho-Akt (A) and phopho-S4 (B); more than 10% of cancer cells were stained and determined as positive staining.
Pharmacokinetics
Everolimus concentrations and pharmacokinetic profiles at the MTD were available in five out of six patients enrolled in the expansion cohort, whereas one patient with early disease progression was excluded as described early. The minimum blood concentration (Cmin), oral clearance (CL/F), and the area under the concentration time curve (AUC0–24) were 7.28 μg l−1, 13.3 l h−1, and 283.5 h*μg l−1, respectively, which were consistent with values in previously reported studies, where everolimus was combined with paclitaxel or docetaxel (Andre et al, 2010; Ramalingam et al, 2010). The maximum blood concentration (Cmax) of everolimus was 24.4 μg l−1 and it was achieved at a median time to Cmax (tmax) of approximately 4 h, with a wide range of 0.98–6.02 h. The interpatient coefficient of variation of pharmacokinetic parameters was less than 40%, except for Cmin with a coefficient of variation of 52%.
Discussion
This phase-1b study provides clinical support for the use of everolimus in combination with paclitaxel for previously treated SCLC patients. It is noteworthy that 5 mg everolimus once daily, which is the level determined as the MTD in the present study, has been previously demonstrated to produce the desired pharmacodynamic effects (Tanaka et al, 2008). Toxicity profiles were tolerable and the overall response rate was 28%. During the whole study period, the most common grade 3–4 adverse events were neutropenia (45%) and thrombocytopenia (15%), which could be managed easily in most cases excepting for one case (neutropenic fever leading to death). In the phase-2 study of single-agent 3-weekly paclitaxel for previously treated SCLC patients, the incidences of grade 3–4 neutropenia and thrombocytopenia were 16% and 8%, respectively (Smit et al, 1998). The observed higher incidence in our study could be attributed to the heavily administered prior therapies or the addition of everolimus. However, taken with the effects of prior therapy on the bone marrow reservoir, the incidence of haematologic adverse events in the present study cannot be regarded as abnormal.
Many trials have evaluated the appropriate schedule of everolimus dosing between daily or weekly administration (O’Donnell et al, 2008; Tanaka et al, 2008; Ellard et al, 2009; Andre et al, 2010). Daily administration inhibited the mTOR pathway more profoundly than did the similar total dose of weekly administered everolimus (Tanaka et al, 2008). In addition, the response rate for 10 mg everolimus daily was 12% compared with 0% for a 70-mg weekly schedule in patients with breast cancer (Ellard et al, 2009). Based on these observations, daily administration has been considered as the appropriate schedule and was adopted in the present study.
In the pharmacokinetic analysis performed at dose level 2, Cmax was lower than anticipated and the delay in tmax was greater than expected. The imprecise tmax can reasonably be attributed to infrequent sampling. However, the chance of a pharmacokinetic interaction between everolimus and paclitaxel might be very low, as it was previously demonstrated that the pharmacokinetic parameters of everolimus were not affected by combined paclitaxel therapy (Campone et al, 2009). It was observed in one study that high-fat meals delayed tmax and reduced Cmax considerably (Kovarik et al, 2002). Our study population was administered with everolimus around 1400 hours after eating lunch on day 1 of cycle 2, when most pharmacokinetic samples were drawn. Therefore, food interaction might have caused the lower Cmax and delayed tmax. Although a modest change in the oral absorption process of everolimus was observed, steady-state systemic exposure was not different from that measured after drug administration under fasting conditions in patients with advanced solid tumours (O’Donnell et al, 2008).
Until now, there has been no molecular determinant to predict the therapeutic responsiveness of everolimus (Rodon et al, 2013). A preclinical study showed that higher expression of phospho-Akt in SCLC cell lines was associated with sensitivity to everolimus (Marinov et al, 2009). On the other hand, in a clinical study, the overexpression of phospho-Akt in baseline tumour tissues was associated with poor overall survival in SCLC patients treated with everolimus, whereas phospho-S6 expression seemed to predict longer disease control duration after everolimus therapy (Tarhini et al, 2010). Although it is limited in number, our data showed no relationship between phospho-Akt or phopho-S6 expression and antitumour effects of everolimus.
In conclusion, 5 mg daily everolimus and 3-weekly paclitaxel 175 mg m−2 could be administered concurrently with manageable toxicity and noticeable responsiveness. However, to increase the clinical benefit of this regimen, the more defined patient selection strategy should be investigated in the further trials.
Change history
17 September 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andre F, Campone M, O’Regan R, Manlius C, Massacesi C, Sahmoud T, Mukhopadhyay P, Soria JC, Naughton M, Hurvitz SA (2010) Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol 28: 5110–5115.
Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN (2012) Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366: 520–529.
Bjornsti MA, Houghton PJ (2004) The TOR pathway: a target for cancer therapy. Nat Rev Cancer 4: 335–348.
Campone M, Levy V, Bourbouloux E, Berton Rigaud D, Bootle D, Dutreix C, Zoellner U, Shand N, Calvo F, Raymond E (2009) Safety and pharmacokinetics of paclitaxel and the oral mTOR inhibitor everolimus in advanced solid tumours. Br J Cancer 100: 315–321.
Dong S, Zhang XC, Cheng H, Zhu JQ, Chen ZH, Zhang YF, Xie Z, Wu YL (2012) Everolimus synergizes with gefitinib in non-small-cell lung cancer cell lines resistant to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Chemother Pharmacol 70: 707–716.
Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, Pritchard K, Eisen A, Vandenberg T, Taylor M, Sauerbrei E, Mishaeli M, Huntsman D, Walsh W, Olivo M, McIntosh L, Seymour L (2009) Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol 27: 4536–4541.
Faried LS, Faried A, Kanuma T, Nakazato T, Tamura T, Kuwano H, Minegishi T (2006) Inhibition of the mammalian target of rapamycin (mTOR) by rapamycin increases chemosensitivity of CaSki cells to paclitaxel. Eur J Cancer 42: 934–947.
Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, Spitznagel EL, Piccirillo J (2006) Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 24: 4539–4544.
Hanna N, Bunn Jr PA, Langer C, Einhorn L, Guthrie T Jr, Beck T, Ansari R, Ellis P, Byrne M, Morrison M, Hariharan S, Wang B, Sandler A (2006) Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J Clin Oncol 24: 2038–2043.
Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB (2002) Inhibition of phosphatidylinositol 3'-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 62: 1087–1092.
Kovarik JM, Hartmann S, Figueiredo J, Rordorf C, Golor G, Lison A, Budde K, Neumayer HH (2002) Effect of food on everolimus absorption: quantification in healthy subjects and a confirmatory screening in patients with renal transplants. Pharmacotherapy 22: 154–159.
Marinov M, Ziogas A, Pardo OE, Tan LT, Dhillon T, Mauri FA, Lane HA, Lemoine NR, Zangemeister-Wittke U, Seckl MJ, Arcaro A (2009) AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res 15: 1277–1287.
Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, Meric-Bernstam F (2004) Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res 10: 7031–7042.
Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, Xiong Y, Zhang S, Krop IE, Winer EP, Kindelberger DW, Coviello J, Sahin AA, Nunez R, Hortobagyi GN, Yu D, Esteva FJ (2011) Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol 29: 3126–3132.
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372: 449–456.
Noda K, Nishiwaki Y, Kawahara M, Negoro S, Sugiura T, Yokoyama A, Fukuoka M, Mori K, Watanabe K, Tamura T, Yamamoto S, Saijo N (2002) Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 346: 85–91.
O’Donnell A, Faivre S, Burris HA III, Rea D, Papadimitrakopoulou V, Shand N, Lane HA, Hazell K, Zoellner U, Kovarik JM, Brock C, Jones S, Raymond E, Judson I (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol 26: 1588–1595.
O’Reilly T, McSheehy PM, Wartmann M, Lassota P, Brandt R, Lane HA (2011) Evaluation of the mTOR inhibitor, everolimus, in combination with cytotoxic antitumor agents using human tumor models in vitro and in vivo. Anticancer Drugs 22: 58–78.
Ramalingam SS, Harvey RD, Saba N, Owonikoko TK, Kauh J, Shin DM, Sun SY, Strychor S, Tighiouart M, Egorin MJ, Fu H, Khuri FR (2010) Phase 1 and pharmacokinetic study of everolimus, a mammalian target of rapamycin inhibitor, in combination with docetaxel for recurrent/refractory nonsmall cell lung cancer. Cancer 116: 3903–3909.
Ren H, Chen M, Yue P, Tao H, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY (2012) The combination of RAD001 and NVP-BKM120 synergistically inhibits the growth of lung cancer in vitro and in vivo. Cancer Lett 325: 139–146.
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10: 143–153.
Schmid K, Bago-Horvath Z, Berger W, Haitel A, Cejka D, Werzowa J, Filipits M, Herberger B, Hayden H, Sieghart W (2010) Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br J Cancer 103: 622–628.
Smit EF, Fokkema E, Biesma B, Groen HJ, Snoek W, Postmus PE (1998) A phase II study of paclitaxel in heavily pretreated patients with small-cell lung cancer. Br J Cancer 77: 347–351.
Sun SY, Fu H, Khuri FR (2006) Targeting mTOR signaling for lung cancer therapy. J Thorac Oncol 1: 109–111.
Tanaka C, O’Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, Raymond E, Zumstein-Mecker S, Stephan C, Boulay A, Hattenberger M, Thomas G, Lane HA (2008) Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol 26: 1596–1602.
Tarhini A, Kotsakis A, Gooding W, Shuai Y, Petro D, Friedland D, Belani CP, Dacic S, Argiris A (2010) Phase II study of everolimus (RAD001) in previously treated small cell lung cancer. Clin Cancer Res 16: 5900–5907.
VanderWeele DJ, Zhou R, Rudin CM (2004) Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther 3: 1605–1613.
Xu CX, Li Y, Yue P, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY (2011) The combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PLoS One 6: e20899.
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Oberg K (2011) Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 364: 514–523.
Acknowledgements
We thank Novartis for their support of this trial.
Author Contributions
JMS, MJA, and KP contributed to the study design. JMS, JL, JSA, MJA, and KP contributed to provision and management of the study patients, data collection, and data interpretation. JMS, JRK, IGD, SYL, YLC, and KP contributed to data analysis and manuscript editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
KP is a consultant for Roche, Eli-Lilly, and Pfizer. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Sun, J., Kim, J., Do, I. et al. A phase-1b study of everolimus plus paclitaxel in patients with small-cell lung cancer. Br J Cancer 109, 1482–1487 (2013). https://doi.org/10.1038/bjc.2013.467
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.467
Keywords
This article is cited by
-
Control of the MYC-eIF4E axis plus mTOR inhibitor treatment in small cell lung cancer
BMC Cancer (2015)
-
Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer
Journal of Cancer Research and Clinical Oncology (2015)
-
SPAG5 upregulation predicts poor prognosis in cervical cancer patients and alters sensitivity to taxol treatment via the mTOR signaling pathway
Cell Death & Disease (2014)
