
Abstract
Background:
Novel prognostic biomarkers and therapeutic strategies are urgently required for malignant melanoma. Ecto-5-prime-nucleotidase (NT5E; CD73) overexpression has been reported in several human cancers. The mechanism(s) underlying deregulated expression and the clinical consequences of changes in expression are not known.
Methods:
We used RT–PCR, qPCR, methylation-specific PCR and pyrosequencing to analyse expression and regulation of NT5E in malignant melanoma cell lines and primary and metastatic melanomas.
Results:
NT5E is subject to epigenetic regulation in melanoma. NT5E mRNA is downregulated by methylation-dependent transcriptional silencing in the melanoma cell lines SKMel2, SKMel23, WM35, Mel501, Mel505 and C81–61 and expression is reactivated by azacytidine. In contrast, the CpG island is unmethylated and the gene expressed in cultured normal melanocytes. In clinical cases of melanoma, methylation in the NT5E CpG island occurs in both primary and metastatic melanomas and correlates with transcriptional downregulation of NT5E mRNA. Relapse with metastatic disease, particularly to the visceral sites and brain, is more common in primary melanomas lacking NT5E methylation. Primary melanomas with methylation in NT5E show limited metastatic potential or more commonly metastasise predominantly to nodal sites rather than viscera and brain (P=0.01).
Conclusion:
Deregulation of NT5E expression in melanoma occurs via epigenetic changes in the NT5E CpG island. Confirmation of our results in larger clinical series would support the candidacy of NT5E as a clinical biomarker in melanoma, which could be applied in both primary and relapsed disease. Inhibition of NT5E may have therapeutic potential in melanoma, particularly in patients with more aggressive disease metastatic to viscera or the brain.
Similar content being viewed by others

Main
Melanoma is an increasingly common, aggressive neoplastic disorder of melanocytes, with a remarkable propensity to disseminate and form metastases in distant organs sites, including the bone, lungs, liver and brain. To date, no adjuvant therapy after control of the primary tumour has shown clear benefit for patients with high-risk primary melanoma and the prognosis for patients with metastatic disease, particularly in visceral sites and brain is frequently very poor. Despite empirical testing of numerous chemotherapeutic regimens and targeted therapies, melanoma remains an essentially incurable disorder in patients with disseminated disease, with survival typically of a few months. Early detection and effective treatment of melanoma at early stages are urgently required, particularly as the incidence of the disease increases and the demography relentlessly moves towards a young patient group. The unique biology of the melanocyte and highly individual pathobiology of melanoma implies that a large number of genes may incur genetic or epigenetic change during the development of melanoma.
Epigenetics is the study of changes in gene expression without concomitant changes in gene structure and is now well recognised as an important mechanism by which the expression of genes may be modified in cancer (Lopez et al, 2009). Although the genome undergoes a process of global hypomethylation during tumorigenesis, specific CpG islands become hypermethylated with associated silencing of genes. Such methylation-dependent transcriptional silencing is a common finding in many human tumours and genes subject to this process are usually tumour suppressors. Methylation of the majority of genes is specific to neoplasia. Furthermore, methylated genomic DNA is stable and the technology for detection of methylated genomic DNA is sensitive and readily amenable to automation. Taken together, these are attractive properties for the use of methylated genomic DNA as a disease biomarker, provided markers of sufficient specificity for routine clinical use can be identified and their utility verified in large patient populations.
Ecto-5-prime-nucleotidase (NT5E; CD73) has a number of documented functions, the best characterised of which is catalysis of the conversion of extracellular purine 5′ mononucleotides to membrane-permeable nucleosides, the preferred substrate being AMP, resulting in local generation of adenosine. Evidence from a number of experimental systems suggests that expression of NT5E may be important in increasing the invasive and metastatic properties of some cancer cells and overexpression of NT5E may contribute to progression of cancer via generation of adenosine (Wang et al, 2011). Further, NT5E-dependent production of adenosine by cancer exosomes causes suppression of T-lymphocyte function (Clayton et al, 2011). Although these observations all support an oncogenic role for NT5E, other properties may be consistent with a potential tumour suppressor function. For example, knockdown of NT5E increases cell migration in specific cell types (Andrade et al, 2011). NT5E is overexpressed in some human tumours, but the mechanism(s) causing overexpression have not been defined. In the present study, we show that NT5E (CD73) is subject to methylation-dependent transcriptional silencing in melanoma, with potential clinical and therapeutic implications.
Materials and Methods
Cell lines
Melanoma cell lines were routinely grown in Dulbecco's modified Eagles's medium or Roswell Park Memorial Institute medium, supplemented with 10% fetal bovine serum. Primary, normal human melanocytes were purchased from Clonetics (Lonza Group, Basel, Switzerland) and grown according to the manufacturer's instructions. All cells were incubated at 37 °C and 5% CO2, with regular mycoplasma contamination test. The characteristics of the cell lines are summarised in Table 1.
Demethylation
Demethylating treatment was as described previously (Lee et al, 2010). Briefly, cells were treated with 5-μ M azacytidine (AZA; Sigma, Gillingham, UK) for 7 days. Cells were split every 2–3 days with the addition of fresh drug. After drug treatment, cells were harvested for qPCR.
Tissues
The study received approval from the Local Research Ethical Committee, East London and City Health Authority and the Tayside Tissue Bank under delegated authority from the Tayside Local Research Ethics Committee. Tissues were obtained under written, informed consent. Melanomas and control benign nevi were from the Pathology archives of Bart's and the London NHS Trust and from the Tayside Skin Cancer Tissue Bank. All tissues underwent histopathological review before inclusion in the study to (i) confirm the diagnosis of melanoma; (ii) to confirm Breslow thickness (BT); and (iii) to verify sufficient representation of neoplastic cells. Melanomas were both primary, cutaneous melanomas and metastatic lesions. Surgical excision was performed according to routine protocols of clinical care. BT and melanoma sub-type is shown in Figure 4 for the primary melanomas. Recurrent melanomas were typically from lymph node resections, (where performed) sentinel lymph node biopsies or cutaneous recurrences. Benign nevi were from surgical excisions and confirmed by histopathological analysis to contain no neoplastic cells.
Analysis of methylation
Genomic DNA was obtained from formalin-fixed paraffin-embedded tissue sections by extended digestion in Proteinase K followed by phenol extraction. Genomic DNA from cell lines and tissue samples was subjected to bisulphite modification using the EZ DNA Methylation Kit (Zymo, Irvine, CA, USA) according to the manufacture's instructions. Methylation in the CpG island of the NT5E gene was analysed with pyrosequencing technology, in which the degree of methylation at each CpG position in a sequence is determined from the ratio of T and C, following bisulphite modification. Primer sequences were as follows: forward 5′-GTATTAGGGTATTATTTGGTTTAT-3′ reverse 5′-BIOT-CTTACCACACTCTACCATCC-3′ fragment size: 170 bp.
Polymerase chain reaction conditions were as follows: 95 °C for 10 min, 95 °C for 30 s/54 °C for 30 s/72 °C for 40 s for 40 cycles, 72 °C for 7 min. Polymerase chain reaction products were resolved through 2% agarose gels, visualised using a transilluminator, then analysed by pyrosequencing using the Biotage Sample Prep kit and using the forward primer for sequencing. Analysis of percentage methylation at each CpG dinucleotide was performed using CpG Software (Qiagen, Crawley, UK). Placental DNA was used as negative control of methylation (0% average methylation) and a commercial methylated DNA (Millipore, Watford, UK) was used as positive control (98% average methylation). Methylation was also analysed by methylation-specific PCR (MSP). DNA (0.5 μg) was modified by sodium bisulphite using the Zymo EZ DNA Methylation kit (Zymo). Bisulfite-modified DNA was used as a template for PCR with primers specific for methylated or unmethylated alleles. CpGenome Universal Methylated DNA (Chemicon Europe, Chandlers Ford, Hampshire, UK) and normal human unmethylated DNA were used as positive and negative controls, respectively, in each experiment. MSP primer sequences: M forward primer: 5′-TATTTTATGAACGTTTTGCGTTACG-3′ M reverse primer: 5′-CTAAACTTACCACACTCTACCATCCG-3′ U forward primer: 5′-ATTTTATGAATGTTTTGTGTTATGA-3′ U reverse primer: 5′-AACTTACCACACTCTACCATCCACT-3′.
Gene expression
For qPCR analysis of expression, total RNA was isolated musing the Recover All Total Nucleic Acid Isolation (Ambion, Life Technologies Italia, Monza, Italy). In all, 25 μl PCR reactions were performed using 50 ng of cDNA obtained by reverse transcription. Amplification and analysis were done according to the manufacturer's protocol in 96-well plates in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Life Technologies Italia, Monza, Italy) and the pre-cast ‘TaqMan Gene Expression Assays’ (Applera, https://products.appliedbiosystems.com/) for NT5E (Hs001573922_m1). Quantification of the target transcript was performed in comparison to the reference transcript β2microglobulin (Hs99999907_m1), using the ‘delta-delta Ct method for comparing relative expression results in real-time PCR as outlined by PE Applied Biosystems (Perkin Elmer, Forster City, CA, USA).
Statistics
NT5E CpG island methylation status and presence or types of metastasis were assessed for associations with the Fisher's exact test. All of the statistical analyses were performed using Prism 5 (GraphPad software, Inc., La Jolla, CA, USA).
Results
NT5E expression is deregulated in highly metastatic C8161 melanoma cells by loss of CpG island methylation from the parental C81–61 cells
We analysed expression of NT5E mRNA in the paired malignant melanoma cell lines C81–61 (low metastatic potential) and the highly metastatic isogenic derivative C8161 (high metastatic potential). We also analysed expression in three additional melanoma cell lines and in normal human melanocytes. NT5E mRNA was abundantly expressed in normal melanoyctes, in C8161 cells and in each of the three other melanoma cell lines. However, expression was not detectable by RT–PCR in C81–61 cells (Figure 1A). A CpG island is located at the 5′ end of the NT5E gene (Figure 1B). To test whether aberrant methylation might be the basis for the low expression of NT5E mRNA in C81–61, we performed bisulphite sequence analysis of the CpG island in C81–61 (absent expression) and the C8161 derivative (high expression). The CpG island was entirely unmethylated in C8161 but densely methylated in C81–61, consistent with aberrant methylation underlying the absence of expression (Figure 1B). On the basis of these observations, we designed primers for MSP and analysed methylation in the cell lines. Methylation was only observed in C81–61 consistent with bisulphite sequencing and absence of expression in this cell line (Figure 1A). To further verify that methylation is the mechanistic basis for NT5E downregulation, we treated C81–61 and C8161 cells with the demethylating agent AZA and measured mRNA levels. NT5E mRNA was increased by AZA in C81–61 but not in C8161 (Figure 1C).

NT5E expression is silenced by methylation in C81–61 cells, but deregulated in highly metastatic isogenic C8161 cells. (A) Expression of NT5E correlates inversely with CpG methylation in C81–61 and C8161. The upper panels are RT–PCR analysis of NT5E and the control gene GAPDH in five melanoma cell lines and normal human melanocytes (NHEM) as indicated. NT5E mRNA is expressed abundantly in all cell lines except C81–61. Lower panels are MSP analysis of the NT5E CpG island in the same cell lines. There is complete methylation in C81–61 but no detectable methylation in C8161. (B) Bisulphite sequencing analysis of the NT5E CpG island in C81–61 and C8161 cell lines. Bisulphite sequencing was performed as described in Materials and Methods and the figure shows a diagrammatic representation of the NT5E CpG island. CpG sites are shown as vertical lines. Methylated CpG dinucleotides are shown as black blocks, unmethylated CpGs as open blocks. Five levels of methylation are indicated: 0-no black blocks; 1–25%-1 black block; 25–50%-2 black blocks; 50–75%-3 black blocks; 75–100%-4 black blocks. Positions of the MSP and bisulphite-sequencing primers are indicated. The CpG island is almost completely methylated in C81–61 cells and entirely unmethylated in C8161, consistent with expression of NT5E mRNA in each cell line. (C) Expression of NT5E mRNA is reactivated by demethylation. Exponentially growing C81–61, C8161 and Mel501 cells were treated with 5′azacytidine (5′ AZA) (black blocks) or untreated (open blocks). cDNA was prepared and expression of NT5E mRNA determined as described in Materials and Methods.
NT5E is frequently methylated in melanoma cell lines
We next analysed NT5E expression in a panel of additional melanoma cell lines. By RT–PCR, NT5E mRNA was expressed in the majority of the cell lines analysed but was undetectable in Mel501 and present only at low levels in Mel505 and PMWK (Figure 2A). To validate these observations, we performed analysis of NT5E expression and methylation in an independent panel of melanoma cell lines (which contained a subset of the lines analysed by semi-quantitative methods) using qPCR and pyrosequencing. Representative pyrograms are shown in Figure 3. In total, there was methylation in 6/16 cell lines (C81–61 (bisulphite sequencing), SKMel2, SKMel23, WM-35, Mel501 and Mel505; Table 1; Figure 2B). There was a good correlation between the presence of methylation in the NT5E CpG island and absent or reduced expression of the mRNA (Figure 2C and D). As in C81–61 cells, demethylation by exposure to AZA caused an increase in NT5E mRNA levels in Mel501 cells (Figure 1C).

Analysis of NT5E expression and methylation in malignant melanoma. (A) Expression of NT5E correlates inversely with CpG methylation in melanoma cell lines. The upper panels are RT–PCR analysis of NT5E and the control gene GAPDH in melanoma cell lines as indicated. Lower panels are MSP analysis of the NT5E CpG island in the same cell lines. There is complete methylation in Mel501 and partial methylation in Mel505. (B) Pyrosequencing analysis of the NT5E CpG island in a panel of melanoma cell lines. Pyrosequencing was done as described in Materials and Methods. The level of methylation is represented by the intensity of shading in the circles, each of which represents an individual CpG dinucleotide in the amplified fragment. The mean % CpG methylation in the amplified fragment is also shown. (C) qPCR analysis of NT5E mRNA levels in panel of melanoma cell lines. Data shown are means of at least three duplicates±1 s.d.

Pyrosequencing analysis of the NT5E CpG island in WM35 and SKMel173 cell lines. The figure shows representative pyrograms. The shaded C residue is the control for bisulphite conversion: there must be no peak at this residue (non CpG cytosine/guanine), confirming 0% cytosine incorporation. % Methylation at each CpG is indicated.
The NT5E CpG island is methylated in clinical cases of melanoma
These results show that expression of NT5E is regulated epigenetically in melanoma cell lines and prompted us to test whether the NT5E CpG island is methylated in vivo. We first analysed, using MSP, CpG methylation in a series of 52 unselected, histologically confirmed melanomas. Consistent with studies in cell lines, there was methylation in the NT5E CpG island in 22/52 (42%) biopsies, as detected by MSP. We validated the frequency of methylation by performing pyrosequencing on a subset of the cases analysed by MSP and this showed a concordance in 8/9 melanomas.
Methylation of NT5E is associated with specific sites of metastasis
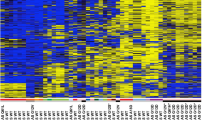
We further investigated methylation in clinical cases of melanoma by analysis of a well-characterised series of primary and metastatic melanomas using pyrosequencing. As controls, we first tested a series of benign nevi. This revealed a mean % methylation of 8.3% in the nevi. Accordingly, we designated a value of 9% as the cut off for methylation in melanomas and proceeded to analyse NT5E CpG methylation in 27 primary melanomas from various sun-exposed sites (Figure 4). Methylation was detected in 8/27 cases (29%), a frequency comparable to the initial series of cases analysed by MSP. Methylation was more common in nonrelapsing cases, although this did not reach statistical significance because of the small number of cases (6/17 vs 2/10; P=0.23). In the 10 cases with subsequent metastatic relapse, five were in visceral sites (the brain, liver, lung and parotid) and five in nonvisceral sites (Figure 4). The NT5E CpG island was methylated in 0/5 cases with visceral metastasis, whereas 2/5 cases with nonvisceral metastasis were methylated in the NT5E CpG island (P=0.16). We noted that the two relapsing cases with methylation were both lymph node metastases.

Methylation in the NT5E CpG island in primary malignant melanoma. Pyrosequencing analysis of the NT5E CpG island in primary malignant melanomas from sun-exposed sites was done as described in Materials and Methods. The level of methylation is represented by the intensity of shading in the circles, each of which represents an individual CpG dinucleotide in the amplified fragment. The mean % CpG methylation in the amplified fragment is also shown. The anatomical site of the primary lesion is indicated, together with the Breslow thickness (BT) in mm and the melanoma type (where available): NMM=nodular malignant melanoma; MM=malignant melanoma, sub-type not available; SSMM=superficial spreading malignant melanoma. Site(s) of metastasis (if applicable) are indicated: Br=brain; Cu=cutaneous; Li=liver; LN=lymph Node; Lu=lung; Or=orbit; Pa=parotid gland; Sp=spleen.
NT5E methylation protects against metastasis in high-risk primary melanomas
The 27 initially analysed cases included 17 with a BT >2 mm, of which 8/17 had relapsed at the time of censor. Relapse was more common in cases lacking NT5E methylation (1/8 methylated vs 7/8 unmethylated), although again because of the small number of cases this difference did not reach statistical significance (P=0.13).
NT5E methylation in nodal/cutaneous metastases predicts risk of visceral metastases
These results prompted us to further examine the apparent inverse correlation between methylation in NT5E and visceral metastasis. We performed pyrosequencing in 26 metastatic lesions from LN or cutaneous sites confirmed by histopathology to be malignant melanoma, which arose in patients with a previous primary melanoma (Figure 5). The NT5E CpG island was methylated in 6/26 cases, using the methylation cutoff of 9% previously established in benign nevi. Metastasis to visceral sites, including bladder, brain, lung and liver, occurred in 12 patients and in all 12 the NT5E CpG island in the sampled lymph node/cutaneous metastasis was unmethylated (P=0.01, OR=19; Figure 5).

NT5E CpG methylation in cutaneous lesions or LN predicts visceral vs nonvisceral sites of distant metastasis. Pyrosequencing was performed as described in Materials and Methods. The level of methylation is represented by the intensity of shading in the circles, each of which represents an individual CpG dinucleotide in the amplified fragment. The mean % methylation is also shown. The site of biopsy is indicated, together with the site of metastasis: Bl, Bo, Br, Cu, Li, LN, Lu, Or, Pa and ST. Also shown (where available) is the Breslow thickness (BT) of the primary melanoma from which the metastatic lesions derived. The superscripts1,2,3 denote paired cutaneous biopsies from the same patients. Note that in each case with paired biopsies, the level of methylation is similar in the two biopsies. Abbreviations: Bl=bladder; Bo=bone; Br=brain; Cu=cutaneous; Li=liver; LN=lymph node; Lu=lung; NA=not available; Or=orbit; ST=soft tissue.
Discussion
Despite advances in understanding of the fundamental biology of the melanocyte and development of therapeutic agents, including the BRAF inhibitor Vemurafenib and the monoclonal antibody Ipilimumab, metastatic malignant melanoma remains an incurable disease with a poor prognosis. As such, additional mechanistic insights to growth and progression of melanoma and novel therapeutic strategies are clearly required. Prognosis at diagnosis of patients with primary melanomas is estimated largely by histopathological parameters, such as BT, presence of ulceration and lympho-vascular invasion, but the ability of these to accurately predict clinical outcomes is limited. As such, molecular genetic biomarkers would be valuable in assessing risk of metastatic relapse to viscera and/or brain: events that are associated with poor clinical outcomes. Similarly, initial relapse in cutaneous or nodal sites is common. Such relapses are typically managed by surgical excision. This is not only therapeutic but provides, as part of routine clinical management a source of tissue for assessment of prognosis, assuming robust biomarkers to predict the risk of later metastatic relapse to viscera and/or brain can be identified. Such biomarkers would be immensely useful to inform clinical management of such patients. Here, we report that expression of NT5E (CD73) is regulated epigenetically in malignant melanoma. We show that expression is downregulated by aberrant methylation in the CpG island located in the 5′ regulatory region of the NT5E gene in melanoma cell lines and clinical melanomas, and we present preliminary evidence that methylation in the NT5E CpG island may have utility as a biomarker of risk of visceral/brain metastasis, both in primary melanoma and in cutaneous and/or nodal recurrence. To the best of our knowledge, this is the first report of epigenetic regulation of this gene in human neoplasia, although a single study has identified upregulation of NT5E by chemical demethylation in a gastric carcinoma cell line, consistent with methylation-dependent transcriptional silencing (Yamashita et al, 2006).
In the initial studies, we showed that NT5E mRNA was abundantly overexpressed in C8161 cells, which have a highly invasive and metastatic phenotype, but was not expressed in (isogenic) parental C81–61 cells, implying that deregulation of NT5E contributes to the greater metastatic potential of C8161 cells. Bisulphite sequencing and MSP revealed that the NT5E CpG island was fully methylated in parental C81–61 cells but was unmethylated in C8161. Taken together with reactivation of expression by AZA, these findings imply that loss of methylation is the mechanistic basis for deregulation of NT5E in C8161 cells. These results not only support a role for NT5E in the more metastatic phenotype of C8161 cells but are also consistent, in a wider context, with mechanistic models in which epigenetic changes in key genes contribute to malignant progression. More commonly in neoplasia, methylation increases in the CpG islands of genes as tumour suppressors are silenced and cancer cells acquire progressively more malignant properties (Lopez et al, 2009). NT5E, however, appears to be a gene in which absence of methylation contributes to a more malignant phenotype. Previous studies on the expression of NT5E have reported overexpression of the gene in invasive melanoma cell lines (Sadej et al, 2006a, 2006b). It was, therefore, unexpected to find that the gene was silenced by methylation in some melanoma cell lines. The selective pressure favouring downregulation of NT5E in some cell lines is not clear. However, it has been previously shown that NT5E mRNA is upregulated during differentiation of melanoma cell lines (Kim et al, 2006). Further, knock-down of NT5E causes increased cell migration in some cell types (Andrade et al, 2011). It is therefore possible that, perhaps in early disease, methylation-dependent silencing of NT5E might block differentiation and/or promote migration in a subset of melanomas.
The studies of clinical melanomas revealed a frequency of NT5E CpG methylation similar to cell lines, using MSP and pyrosequencing. We show that methylation in both primary melanomas and in cutaneous and lymph node metastases influences the clinical behaviour of the disease, cases with methylation being less likely to metastasise to visceral sites such as the lung and liver and brain, whereas cases with methylation more commonly metastasise to nodal sites. Of particular interest, in patients with primary melanomas of BT >2 mm, which are at high increased risk of metastasis, methylation in the NT5E CpG island was associated with a lower risk of metastasis to visceral sites and brain than cases lacking NT5E methylation. The potential biomarker utility of NT5E methylation was also evident in relapsed/metastatic lesions from cutaneous or nodal sites, cases with NT5E methylation being more likely to subsequently metastasise to viscera or brain if the NT5E CpG island was unmethylated. Such nonvisceral metastatic lesions are typically treated as part of routine clinical management by surgical resection and tissue obtained from these procedures would clearly be amenable to analysis of biomarkers such as NT5E. As normal stromal skin tissue also expresses high levels of NT5E (http://www.proteinatlas.org/ENSG00000135318), immunohistochemical analysis of NT5E may not be prognostically informative. Importantly, we have shown in the present study that NT5E methylation levels are low in normal melanocytes and benign nevi, emphasising the specificity of NT5E methylation for neoplastic melanocytes. In some cases, surgically excised melanomas contain a significant normal tissue component, potentially limiting the biomarker utility of detection of NT5E CpG methylation, but this could be readily overcome by micro-dissection of tissue sections before methylation analysis. The sensitivity of methylation analysis and its specificity for neoplasia implies that NT5E methylation may be more informative than immunohistochemistry as a prognostic biomarker. Studies to address this question are in progress.
Our results are consistent with animal models in which NT5E overexpression is associated with aggressive behaviour of cancers (Stagg et al, 2011) and in mouse models in which the growth of primary tumours and formation of metastases are reduced in mice lacking NT5E (Yegutkin et al, 2011). Aside from the potential biomarker utility of NT5E, the likely role of NT5E in driving metastatic disease in a proportion of cases implies that NT5E may be a viable therapeutic target in melanoma, particularly in clinically challenging cases with visceral and/or brain metastases. Evidence from animal models suggests that inhibition of both host and tumour cell NT5E has anti-cancer effects (Wang et al, 2011; Yegutkin et al, 2011), emphasising the likely value of inhibition of NT5E. Our results suggest that use of NT5E inhibitors should be informed, at least in part, by analysis of the methylation status of the CpG island.
In conclusion, we show that methylation in the NT5E CpG island is an important determinant of metastatic potential of malignant melanoma. Confirmation of these results in larger, independent series would support the candidacy of NT5E as a potentially clinically useful epigenetic biomarker to inform management of melanoma patients, particularly those with high-risk primary lesions and in those with nodal and/or cutaneous recurrences/metastases.
Change history
23 January 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andrade CMB, Lopez PLC, Noronha BT, Wink MR, Borojevic R, Margis R, Lenz G, Battastini AMO, Guma FCR (2011) Ecto-5′-nucleotidase/CD73 knockdown increases cell migration and mRNA level of collagen I in a hepatic stellate cell line. Cell Tissue Res 344: 279–286
Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z (2011) Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol 187: 676–683
Kim G, Kim D-J, Lee S-H, Yeom Y-I, Kang D (2006) Gene expression profile during Mezerein-induced reversible differentiation of human melanoma cells. Kor J Gerontol 16: 201–210
Lee S, Syed N, Taylor J, Smith P, Griffin B, Baens M, Bai M, Bourantas K, Stebbing J, Naresh K, Nelson M, Tuthill M, Bower M, Hatzimichael E, Crook T (2010) DUSP16 is an epigenetically-regulated determinant of JNK signalling in Burkitt lymphoma. Br J Cancer 103: 265–274
Lopez J, Percharde M, Coley HM, Webb A, Crook T (2009) The context and potential of epigenetics in oncology. Br J Cancer 100: 571–577
Sadej R, Spychala J, Skladanowski AC (2006a) Ecto-5′-nucleotidase (eN, CD73) is co-expressed with metastasis promoting antigens in human melanoma cells. Nucleosides Nucleotides Nucleic Acids 25: 1119–1123
Sadej R, Spychala J, Skladanowski AC (2006b) Expression of ecto-5′-nucleotidase (eN, CD73) in cell lines from various stages of human melanoma. Melanoma Res 16: 213–222
Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, Smyth MJ (2011) CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 71: 2892–2900
Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, Zhang B (2011) CD73 has distinct roles in non hematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 121: 2371–2382
Yamashita S, Tsujino Y, Moriguchi K, Tatematsu M, Ushijima T (2006) Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2′-deoxycytidine treatment and oligonucleotide micro-array. Cancer Sci 97: 64–71
Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemelä J, Laurila JP, Elima K, Jalkanen S, Salmi M (2011) Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol 41: 1231–1241
Acknowledgements
The study was supported by Barts and the London Charity, The Brain Tumour Research Charity (BTRC), The Leng Foundation, The Medical Research Council and Tayside Tissue Bank. Eleftheria Hatzimichael is a scholar of The Hellenic Society of Haematology Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wang, H., Lee, S., Nigro, C. et al. NT5E (CD73) is epigenetically regulated in malignant melanoma and associated with metastatic site specificity. Br J Cancer 106, 1446–1452 (2012). https://doi.org/10.1038/bjc.2012.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.95
Keywords
This article is cited by
-
Prognostic significance of NT5E/CD73 in neuroblastoma and its function in CSC stemness maintenance
Cell Biology and Toxicology (2023)
-
Protean role of epigenetic mechanisms and their impact in regulating the Tregs in TME
Cancer Gene Therapy (2022)
-
The adenosine pathway in immuno-oncology
Nature Reviews Clinical Oncology (2020)
-
CD73 expression on effector T cells sustained by TGF-β facilitates tumor resistance to anti-4-1BB/CD137 therapy
Nature Communications (2019)
-
Inhibition of the adenosinergic pathway: the indispensable part of oncological therapy in the future
Purinergic Signalling (2019)
