
Abstract
Background:
Round 1 data of human papillomavirus (HPV) FOCAL, a three-arm, randomised trial, which aims to establish the efficacy of HPV DNA testing as a primary screen for cervical cancer, are presented.
Methods:
The three arms are: Control arm – liquid based cytology with atypical squamous cells of unknown significance (ASC-US) triage with hrHPV testing; Intervention Arm – hrHPV at entry with liquid-based cytology (LBC) triage of hrHPV positives, with exit screen at 4 years; Safety check arm – hrHPV at entry with LBC triage of hrHPV positives with exit screen at 2 years.
Results:
A total of 6154 women were randomised to the control arm and 12 494 to the HPV arms (intervention and safety check). In the HPV arm, the baseline cervical intraepithelial neoplasia (CIN)2+ and CIN3+ rate was 9.2/1000 (95%CI; 7.4, 10.9) and 4.8/1000 (95%CI; 3.6, 6.1), which increased to 16.1/1000 (95%CI 13.2, 18.9) for CIN2+ and to 8.0/1000 (95%CI; 5.9, 10.0) for CIN3+ after subsequent screening of HPV-DNA-positive/cytology-negative women. Detection rate in the control arm remained unchanged after subsequent screening of ASC-US-positive/hrHPV DNA-negative women at 11.0/1000 for CIN2+ and 5.0/1000 for CIN3+.
Conclusion:
After subsequent screening of women who were either hrHPV positive/cytology negative or ASC-US positive/HPV negative, women randomised to the HPV arms had increased CIN2+ detection compared with women randomised to the cytology arm.
Similar content being viewed by others
Main
Secondary prevention of cervical cancer relies on the accurate diagnosis and treatment of its high-grade precursors, cervical intraepithelial neoplasia (CIN) 2 and CIN3, to prevent progression to invasive disease. There is now well-established evidence demonstrating that cervical infection with high-risk human papillomavirus (HPV) is the cause of cervical cancer (Walboomers et al, 1999), and high-risk HPV DNA (hrHPV) testing of cervical samples substantially improves the sensitivity for detecting relevant precancerous lesions compared with cytology. As well, longitudinal studies have demonstrated that the negative predictive value of an HPV DNA test for CIN3+ or cancer is substantially higher than that of a negative cytology (Dillner et al, 2008). The question remains as to whether implementation of HPV DNA testing as the primary screen as part of a centralised cervical cancer screening programme offers better detection of precancerous lesions, and will thereby ultimately reduce cancer morbidity and mortality, compared with cytology.
Two large European trials have explored the impact of primary hrHPV testing vs cytology in the control arms on incidence of CIN2+ or CIN3+ (Cuzick et al, 2008; Leinonen et al, 2009; Anttila et al, 2010; Ronco et al, 2010). The HPV FOCAL study is the first North American randomised controlled trial comparing HPV testing vs liquid-based cytology (LBC) Pap tests as the primary screen for cervical cancer (Ogilvie et al, 2010). Based in British Columbia, Canada, HPV FOCAL aims to establish the efficacy of HPV DNA testing as a primary screening test with a screening interval of 4 years, in a population of women eligible for screening in a Canadian organised screening programme. In this paper, baseline findings for Round 1 screening results on women recruited before January, 2011 are presented.
Materials and methods
Study design
Study methodology, including description of the trial design, inclusion and exclusion criteria, interventions, outcomes, sample size calculations and randomisation procedures are detailed elsewhere. Briefly, HPV FOCAL is a three-arm, randomised controlled trial (Figure 1) (Ogilvie et al, 2010) and enrolment commenced in January 2008:

Recruitment to the HPV FOCAL Study.
Control arm
Liquid-based cytology at entry and again at 2 years: ⩾low-grade squamous intraepithelial lesion (LSIL) cases referred to colposcopy; atypical squamous cells of unknown significance (ASC-US) cases are triaged with hrHPV testing and those hrHPV negative recalled at 12 months with LBC, those hrHPV positive referred to colposcopy. Exit at 4 years with LBC and hrHPV and referred to colposcopy if positive on either test.
Intervention arm
High-risk HPV at entry with LBC triage of hrHPV positives; immediate referral to colposcopy if LBC positive. If hrHPV positive/LBC negative, recalled at 12 months for hrHPV and LBC testing with referral to colposcopy if positive on either. Exit at 4 years with hrHPV and LBC with referral to colposcopy if positive on either test.
Safety check arm
High-risk HPV at entry with LBC triage of hrHPV positives: immediate referral to colposcopy if LBC positive. If hrHPV positive/LBC negative, recalled at 12 months for hrHPV and LBC testing with referral to colposcopy if positive on either. Exit at 2 years with LBC: ASC-US cases are triaged with hrHPV testing.
Although detailed in an earlier publication (Ogilvie et al, 2010), there have been slight revisions in the trial protocol since that time. Recruitment to the safety arm ceased in December 2010 and randomisation has continued to the remaining two arms. In the original version of the trial protocol, when women in the Safety and Intervention arms were hrHPV positive/LBC negative at baseline, they were initially recalled for hrHPV and LBC at 6 months and if ⩾ASC-US (regardless of hrHPV result) referred to colposcopy at this 6-month visit. This 6-month visit was eliminated from the protocol in March 2010 and women were recalled at 12 months. For the purposes of this paper, referral rates and results are combined for 6- and 12-month visit. In addition, as women in the Safety and Intervention arms are identically managed at baseline and initial follow-up, the results for both of these arms have been combined and referred to as the ‘HPV arm’.
Study population and recruitment
Cervical cancer screening and follow-up in British Columbia is operated through a single organised screening registry, including a provincial cytology laboratory where cervical screens in the province are received and processed, and a coordinated colposcopy programme. Screening history data for all women in the province is maintained in one central provincial database. Women aged 25 to 65 who participate in the provincial programme, and receive care from a trial collaborating family physician (CFP) are eligible to participate in the trial. Eligible women in the CFP practices are sent a centrally generated invitation letter from the CFP office as they approach their scheduled rescreening date (typically 2 years after last screen). Women not receiving an invitation letter but who are participants in the provincial screening programme and eligible to participate in the trial may be identified by the CFP. In British Columbia, family practices are not rostered and women may receive medical services from multiple family practitioners.
Collaborating family physicians and participants are initially blinded to study arm allocation. If the screening results are hrHPV and/or cytology positive, the results are communicated to the CFP along with the recommended follow-up. Negative baseline results are reported as ‘no abnormality detected’, and clinicians are advised that follow-up recommendations will be communicated to them in 2 years, to preserve randomisation allocation.
Screening tests
The study sample is placed in a ThinPrep PreservCyt vial (Hologic Inc, Bedford, MA, USA) and is used for all the HPV FOCAL trial testing. Study arm randomisation occurred at the time of sample receipt by the laboratory. For those allocated to the hrHPV-testing arms, an aliquot is removed and processed using the Qiagen sample conversion kit, and tested using the Digene Hybrid Capture 2 (HC2) assay (Qiagen, Mississauga ON, USA). LBC testing is performed on the ThinPrep 2000 Processor. The HC2 positive cutoff value is ⩾1.0 (signal/cutoff ratio). The HC2-negative cutoff value is <1.0 s/co. The raw data relative light unit signal is captured but not reported. All testing is carried out according to the manufacturers’ recommendations. Cytological evaluation and reporting follow the Bethesda classification system (Solomon et al, 2002). Cytotechnologists and pathologists interpreting LBC are aware of the sample’s inclusion in the study and of the preceding result for reflex tests, for example, when hrHPV positives are reflexed to LBC.
Colposcopy and histology
Women with screening results of ⩾LSIL, ASC-US/hrHPV positive or with persistent hrHPV positive were referred to colposcopy. All colposcopy examinations and management were performed using standard provincial guidelines at two study colposcopy clinics (Vancouver and Victoria). For the purposes of the trial, biopsies are required on all colpscopic procedures. Colposcopists were aware of cytology and hrHPV results to mirror the real-world management of lesions, and each participant referred to colposcopy received either a directed biopsy for lesions or endocervical curettage for histological analysis if no lesion seen. Histopathological interpretation of biopsies is conducted at two laboratories by study pathologists who are blinded to the screening results that led the patient to colposcopy.
Ethical issues
This study is being conducted in accordance with the Ethical Conduct for Research Involving Humans Tri-Council Policy Statement (http://www.pre.ethics.gc.ca/eng/policy-politique/initiatives/tcps2-eptc2/Default/). Ethics approval has been obtained from appropriate local research ethics boards. International Standard Randomized Controlled Trial Number Register is ISRCTN79347302.
Statistical analysis
The analysis was based upon women recruited before January 2011 and reports results on the first round of screening. The data from the safety and intervention arms have been combined, because the first-round testing in the two arms is identical. ‘Baseline screen’ relates to the results of the first testing, including triage (Figures 2 and 3). ‘Subsequent screen’ relates to the testing and results for women who were recommended for subsequent testing at 12 months. ‘Overall screen’ relates to combined baseline screen and subsequent screen results. Detection rates were calculated from the number of screen-detected cervical lesions divided by the total number of women tested. Overall detection rates were calculated from the number of screen-detected cervical lesions per woman at both baseline and subsequent testing. Colposcopy referral rates were calculated in the same manner as detection rates. Not all women included in this analysis had received recommended colposcopy or 12-month subsequent testing, as the trial is still proceeding. In these cases, data were treated as being missing-at-random and rates were based upon available results. A Jack Knife approach was used to calculate rates and confidence intervals for the binomial count data and bootstrap simulation was used to determine 95% confidence intervals for the detection rate ratios and the number needed to treat. Chi-square testing was used to compare categorical demographic data and proportions are tested using a Z-test for proportions.

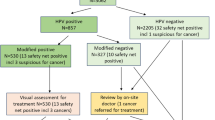
Trial Profile HPV FOCAL – Control Arm. ⩾LSIL: low-grade squamous intraepithelial lesion or higher, including atypia.

Trial profile HPV FOCAL – HPV arm (safety and intervention arm combined). ⩾LSIL: low-grade squamous intraepithelial lesion or higher, including atypia.
Results
By 10 January 2011, the 167 CFPs sent out 44 099 invitations to eligible patients and reminders were sent 4 weeks after the first invitation. In addition, CFPs invited women who met eligibility criteria and presented for cervical cancer screening at their offices to participate in the study. From these two recruitment routes, 18 648 women were randomised in the study, 5271 formally declined participation, 475 did not meet inclusion criteria and 24 086 did not respond to the invitation letter for a total of 29 832 excluded. Of the 18 648 women in the provincial screening programme randomised into the trial, 4381 were identified by the CFP and 14 267 were centrally generated from the BC Cancer Agency through the invitation letter process. In all, 6154 were randomised to the control arm and 12 494 to the HPV arms (intervention and safety check) (Figure 1).
The mean age of trial participants was 46, 47% had a university degree, 37% had ever smoked, 56% reported five or fewer lifetime male sexual partners and most described their cultural background as British (56%), followed by other European (44%). Trial arms were balanced on age, education, ethnic status, smoking and number of partners (Table 1).
Of the 6115 women in the control arm who have completed baseline testing, 5877 (96.1%) were cytology negative (Figure 2). In all, 42 out of 63 (66.6%) women with ASC-US were hrHPV negative, and were advised to return for subsequent testing in 12 months. All other women with abnormal cytology were referred to colposcopy, resulting in a baseline screen referral rate of 32.1/1000 (Table 2). The baseline detection rate in the control arm was 11.0/1000 for CIN2+ and 5.0/1000 for CIN3+ (Table 2). For the 37 ASC-US/hrHPV-negative women for whom subsequent testing had been performed, 31 (83.7%) were cytology negative and returned to regular screening. The remaining six were cytology positive at an ASC-US threshold, and attended colposcopy. Of these, one woman had CIN1 and the remainder had normal pathology results. The overall colposcopy referral rate in the control arm increased slightly after subsequent testing to 33.2/1000 but the CIN2/CIN3-detection rate was essentially unchanged after the subsequent screening in the control arm.
Of the 12 472 women who completed baseline screening in the HPV arm, 11 463 (91.9%) women were hrHPV negative (Figure 3). A total of 648 out of 1009 (64.2%) hrHPV-positive women had normal cytology, and they were recommended for subsequent testing at 12 months. The baseline colposcopy referral rate in the HPV arm was 28.9/1000 (95%CI 26.0, 31.9), similar to the baseline colposcopy referral rate in the control arm (Table 2). The baseline CIN2+ rate was 9.2/1000 (95%CI; 7.4, 10.9) and the baseline CIN3+ rate was 4.8/1000 (95%CI 3.6, 6.1) for the HPV arm (Table 2). To date, there are results for 380 of the 648 women recommended for subsequent testing with both hrHPV and LBC following a baseline hrHPV-positive/cytology-negative result. Women who were either ⩾ASC-US or hrHPV positive in the subsequent round were referred to colposcopy. Only four of the cytology-positive women in subsequent were hrHPV negative, whereas 125 cytology-negative women were hrHPV positive. In the subsequent round of testing, the adjusted colposcopy referral rate was substantially higher, at 544.7/1000 (95%CI; 494.5, 594.9) and the CIN2+ detection rate in the subsequent round was also substantially increased at 244.1/1000 (95%CI; 168.8, 319.4) and 110.2/1000 (95%CI; 55.3, 165.1) for CIN3+. In the HPV arm, the overall detection rates increased from the baseline screening to 16.1/1000 (95%CI 13.2, 18.9) for CIN2+ and to 8.0/1000 (95%CI; 5.9, 10.0) for CIN3+.
The positive predictive value for the HPV arms was initially 31.6% (95%CI: 26.6; 36.6) for CIN2+ and 16.7% (95%CI: 12.7; 20.7) for CIN3+ (Table 2). Overall, the positive predictive value for the HPV arm dropped slightly to 28.1% (95%CI: 23.6, 32.6) for CIN2+ and 13.9% (95%CI: 10.5, 17.3) for CIN3+, after persistently hrHPV-positive women were referred for colposcopy after the subsequent testing round.
Discussion
In this paper, the interim findings of Round 1 for over 18 000 women of a primary HPV screening trial based within an organised cervical screening programme in Canada are presented. At baseline screening, the CIN2+ detection rates were similar in the cytology and HPV arms. However, after subsequent screening with the recommended reflex testing for women who were either persistently hrHPV positive or persistently ASC-US positive, women randomised to the HPV arms had increased CIN2+ detection compared with women randomised to the cytology arm. In contrast, the CIN2+ detection rate remained unchanged after subsequent testing of persistently ASC-US-positive women in the cytology arm.
The potential benefits of using the detection of hrHPV DNA for primary cervical cancer screening are increasingly apparent. However, the high rate of insignificant HPV infections, especially in women under the age of 35 years, has been an impediment to its potential use, so exploring the ability to improve the specificity of hrHPV testing is a crucial step for successful implementation of HPV-based cervical cancer screening (Ronco et al, 2006). In this study, preliminary results indicate that use of cytology triage reduces the number of women who would potentially be referred to colposcopy on the basis of a single hrHPV test. The rates of referral to colposcopy at baseline testing were similar in the HPV and control arms and, as both arms have abnormal cytology, this suggests that similar women are being referred in each arm. Results from the baseline screen of the first round demonstrate that is what happens, with disease detection rates being similar in both arms. However, re-testing of hrHPV-positive women at up to 12 months after baseline screening restores sensitivity, while still managing to control specificity by providing an opportunity for insignificant hrHPV infections to regress. There is the possibility to further increase specificity by extending the re-testing interval for hrHPV-positive/cytology-negative women to 24 months with little potential for disease progression. This may offer a similar disease detection rate to subsequent testing at 12 months, but could decrease unnecessary colposcopies in women by providing more time for transient hrHPV infections to resolve.
In British Columbia, screening participation is not delivered through a population-based invitation framework but relies on physician guidelines and communication between the laboratory and practitioner. Over the past decade, family physician availability and practices have altered and episode-based care has become more common. In addition, women may seek care from multiple sources. Ethical guidelines in Canada require that all women actively consent to participate in the trial, and privacy laws limit direct contact to potential participants for research purposes, that is, invitation letters must come from family physicians willing to participate in the trial. Thus, although 44 000 invitations were sent out by CFPs, the study centre is unable to directly confirm what proportion of women actually received, reviewed and understood their invitation letter. Consequently, this could impact on the representativeness and overall disease risk of women who participate in the trial compared with women who participate in cervical cancer screening in British Columbia. It is certainly accepted that the complex nature of consent required for randomised trials impacts on the general representativeness of participants compared with the overall population, which could impact on overall risk for disease equally in both arms. However, the fundamental question for this study remains the relative effectiveness of the two screening tools, and given the quality of the trial design, we believe the validity of our findings are maintained.
One of the significant concerns with the use of hrHPV testing for primary cervical screening is the increased use of colposcopy services for women who are hrHPV positive. This is a particular concern, both from a systems perspective, with increased cost and unnecessary use of colposcopy, and from an individual perspective, because of the unintended negative consequences of ablative treatment on future fertility and reproductive outcomes (Kyrgiou et al, 2006). Thus, the careful monitoring of colposcopy referral patterns in each arm is essential. In British Columbia, because there is a highly standardised colposcopy programme, this facilitates standardised measurement, with reasonable confidence, the impact of the actual screening tool (hrHPV vs cytology) on referral patterns to colposcopy. Colposcopy referral rates were similar in both the HPV and control arm at baseline screening, but as a result of the follow-up of hrHPV-positive/cytology-negative women, the colposcopy referral rate increased substantially overall.
Although LBC was initially promoted as offering improved sensitivity and disease detection compared with conventional cytology, this has not been consistently supported by the published literature (Ronco et al, 2007; Siebers et al, 2009). Nonetheless, LBC collection is widely used, and offers an important advantage as it supports both hrHPV DNA testing and cytology from a single sample. Authors have noted that one reason for reluctance to switch to hrHPV screening may be that the previous clinical trials comparing hrHPV testing and cytology used conventional Pap smears, as opposed to LBC, and thus the benefits of HPV testing may have been over-estimated (Castle, 2010). The HPV FOCAL study offers an important opportunity to compare hrHPV testing against LBC, and will help to inform this important discussion. Liquid-based cytology also offers other benefits compared with conventional smears for HPV-based primary cervical cancer screening. Women can simply have one specimen taken, and reflex testing can be done on the specimens, avoiding the need for either a repeat visit or dual specimen collection. In addition, liquid-based specimens could provide opportunities for other testing, such as cervical cancer biomarkers and sexually transmitted infections.
The HPV FOCAL study has particular methodological strengths that warrant consideration and position the study to offer key guidance for cervical cancer screening recommendations. This study is based in British Columbia, where cervical cancer screening, follow-up and care are all organised through one centralised provincial agency, the BC Cancer Agency. All Pap smears are read at one centralised laboratory, with high-quality assurance and standardised cytology interpretation and recommendations for care. The HPV FOCAL participants are drawn from a broad base of family practices. For HPV FOCAL, all colposcopies are conducted at BC Cancer Agency referral centres, and colposcopy care is founded in well-established care protocols, offering even further standardisation and minimising heterogeneity in colposcopy interpretation, recommendations and management for all participants in the HPV FOCAL trial. Finally, all histological specimens are analysed at two pathology sites, both of which are experienced cancer referral centres in the province. Pathologists are blinded to the screening results at the time of interpretation of histology specimens. The highly organised nature of screening and post-screening disease diagnosis and management in British Columbia are a particular strength of HPV FOCAL. Recommendations were made for future cervical cancer screening trials to ensure similar delivery of colposcopy, and the HPV FOCAL trial has achieved this owing to the organisational structure of cervical cancer screening in British Columbia (Whitlock et al, 2011).
The protocol of HPV FOCAL is closely aligned with proposed EUROGIN screening algorithm, which outlines the future of cervical cancer screening for developed countries (Franco and Cuzick, 2008; Franceschi et al, 2009). The HPV FOCAL study is well positioned to determine the utility of this proposed approach and to provide evidence for cervical cancer screening guidelines internationally, and further data from completed Round 1 and Round 2 of screening will validate the potential impact of hrhPV on cervical cancer prevention in organised screening programs.
References
Anttila A, Kotaniemi-Talonen L, Leinonen M, Hakama M, Laurila P, Tarkkanen J, Malila N, Nieminen P (2010) Rate of cervical cancer, severe intraepithelial neoplasia, and adenocarcinoma in situ in primary HPV DNA screening with cytology triage: randomised study within organised screening programme. BMJ 340: c1804
Castle PE (2010) Screening: HPV testing for cervical cancer: the good, the bad, and the ugly. Nat Rev Clin Oncol 7: 364–365
Cuzick J, Arbyn M, Sankaranarayanan R, Tsu V, Ronco G, Mayrand MH, Dillner J, Meijer CJ (2008) Overview of human papillomavirus-based and other novel options for cervical cancer screening in developed and developing countries. Vaccine 26 (Suppl 10): K29–K41
Dillner J, Rebolj M, Birembaut P, Petry KU, Szarewski A, Munk C, de Sanjose S, Naucler P, Lloveras B, Kjaer S, Cuzick J, van Ballegooijen M, Clavel C, Iftner T Joint European CS (2008) Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 337: a1754
Franceschi S, Cuzick J, Herrero R, Dillner J, Wheeler CM (2009) EUROGIN 2008 roadmap on cervical cancer prevention. Int J Cancer 125: 2246–2255
Franco EL, Cuzick J (2008) Cervical cancer screening following prophylactic human papillomavirus vaccination. Vaccine 2008: 16–23
Kyrgiou M, Koliopoulos G, Martin-Hirsch P, Arbyn M, Prendiville W, Paraskevaidis E (2006) Obstetric outcomes after conservative treatment for intraepithelial or early invasive cervical lesions: systematic review and meta-analysis. Lancet 367: 489–498
Leinonen M, Nieminen P, Kotaniemi-Talonen L, Malila N, Tarkkanen J, Laurila P, Anttila A (2009) Age-specific evaluation of primary human papillomavirus screening vs conventional cytology in a randomized setting. J Natl Cancer Inst 101: 1612–1623
Ogilvie GS, van Niekerk DJ, Krajden M, Martin RE, Ehlen TG, Ceballos K, Peacock SJ, Smith LW, Kan L, Cook DA, Mei W, Stuart GC, Franco EL, Coldman AJ (2010) A randomized controlled trial of Human Papillomavirus (HPV) testing for cervical cancer screening: trial design and preliminary results (HPV FOCAL Trial). BMC Cancer 10: 111
Ronco G, Cuzick J, Pierotti P, Cariaggi MP, Dalla PP, Naldoni C, Ghiringhello B, Giorgi-Rossi P, Minucci D, Parisio F, Pojer A, Schiboni ML, Sintoni C, Zorzi M, Segnan N, Confortini M (2007) Accuracy of liquid based versus conventional cytology: overall results of new technologies for cervical cancer screening: randomised controlled trial. [see comment]. BMJ 335: 28
Ronco G, Giorgi-Rossi P, Carozzi F, Confortini M, Dalla PP, Del Mistro A, Ghiringhello B, Girlando S, Gillio-Tos A, De Marco L, Naldoni C, Pierotti P, Rizzolo R, Schincaglia P, Zorzi M, Zappa M, Segnan N, Cuzick J (2010) Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomised controlled trial. Lancet Oncol 11: 249–257
Ronco G, Giorgi-Rossi P, Carozzi F, Dalla PP, Del Mistro A, De Marco L, De Lillo M, Naldoni C, Pierotti P, Rizzolo R, Segnan N, Schincaglia P, Zorzi M, Confortini M, Cuzick J New Technologies for Cervical Cancer Screening Working Group New Technologies for Cervical Cancer Screening Working Group (2006) Human papillomavirus testing and liquid-based cytology in primary screening of women younger than 35 years: results at recruitment for a randomised controlled trial. Lancet Oncol 7: 547–555
Siebers AG, Klinkhamer PJJM, Grefte JMM, Massuger LFAG, Vedder JEM, Beijers-Broos A, Bulten J, Arbyn M (2009) Comparison of liquid-based cytology with conventional cytology for detection of cervical cancer precursors: a randomized trial. JAMA 302: 1757–1764
Solomon D, Davey D, Kurman R, Moriarty A, O'Connor D, Prey M, Raab S, Sherman M, Wilbur D, Wright T, Young N Forum Group, Bethesda 2001 Workshop (2002) The 2001 Bethesda System: terminology for reporting results of cervical cytology. JAMA 287: 2114–2119
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N (1999) Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189: 12–19
Whitlock EP, Vesco KK, Eder M, Lin JS, Senger CA, Burda BU (2011) Liquid-based cytology and Human Papillomavirus testing to screen for cervical cancer: a systematic review for the US Preventive Services Task Force. Ann Intern Med 155 (10): 687–697,, W214-5
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
GSO, MK, DVK, REM, TGE, KC, LWS, LK, DAC, SP, GCES, AJC do not report any conflicts of interest relevant to this study. ELF declares no conflict of interest but he has occasionally served as consultant or advisory board member to companies involved with cervical cancer screening (Ikonisys, Cytyc) and HPV diagnostics (Roche, Gen-Probe, BD).
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ogilvie, G., Krajden, M., van Niekerk, D. et al. Primary cervical cancer screening with HPV testing compared with liquid-based cytology: results of round 1 of a randomised controlled trial – the HPV FOCAL Study. Br J Cancer 107, 1917–1924 (2012). https://doi.org/10.1038/bjc.2012.489
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.489
Keywords
This article is cited by
-
Cervical cancer screening program in Indonesia: is it time for HPV-DNA tests? Results of a qualitative study exploring the stakeholders’ perspectives
BMC Women's Health (2024)
-
Correlates of women’s intentions to be screened for human papillomavirus for cervical cancer screening with an extended interval
BMC Public Health (2016)
-
Disease detection and resource use in the safety and control arms of the HPV FOCAL cervical cancer screening trial
British Journal of Cancer (2016)
-
Comparison of the Roche cobas® 4800 and Digene Hybrid Capture® 2 HPV tests for primary cervical cancer screening in the HPV FOCAL trial
BMC Cancer (2015)
-
Cost Effectiveness of Human Papillomavirus-16/18 Genotyping in Cervical Cancer Screening
Applied Health Economics and Health Policy (2015)
