
Abstract
Background:
The controversies concerning possible overtreatment of prostate cancer, highlighted by debate over PSA screening, have highlighted active surveillance (AS) as an alternative management option for appropriate men. Regional differences in the underlying prevalence of PSA testing may alter the pre-test probability for high-risk disease, which can potentially interfere with the performance of selection criteria for AS. In a multicentre study from three different countries, we examine men who were initially suitable for AS according to the Toronto and Prostate Cancer Research International: Active Surveillance (PRIAS) criteria, that underwent radical prostatectomy (RP) in regards to:1.the proportion of pathological reclassification(Gleason score ⩾7, ⩾pT3 disease),2.predictors of high-risk disease,3.create a predictive model to assist with selection of men suitable for AS.
Methods:
From three centres in the United Kingdom, Canada and Australia, data on men who underwent RP were retrospectively reviewed (n=2329). Multivariable logistic regression was performed to identify predictors of high-risk disease. A nomogram was generated by logistic regression analysis, and performance characterised by receiver operating characteristic curves.
Results:
For men suitable for AS according to the Toronto (n=800) and PRIAS (410) criteria, the rates for upgrading were 50.6, 42.7%, and upstaging 17.6, 12.4%, respectively. Significant predictors of high-risk disease were:•Toronto criteria: increasing age, cT2 disease, centre of diagnosis and number of positive cores.•PRIAS criteria: increasing PSA and cT2 disease.Cambridge had a high pT3a rate (26 vs 12%). To assist selection of men in the United Kingdom for AS, from the Cambridge data, we generated a nomogram predicting high-risk features in patients who meet the Toronto criteria (AUC of 0.72).
Conclusion:
The proportion of pathological reclassification in our cohort was higher than previously reported. Care must be used when applying the AS criteria generated from one population to another. With more stringent selection criteria, there is less reclassification but also fewer men who may benefit from AS.
Similar content being viewed by others


Main
Treatment paradigms for small-volume, low-grade prostate cancer (PCa) are currently moving away from radical treatments. The recent controversial US preventative task force report (Lin et al, 2011) giving PSA screening a ‘D’ rating, prompted by the results from the PLCO and ERSP trials, has highlighted the issues associated with overdiagnosis and overtreatment of PCa. In follow-up of over 3,500 patients across seven active surveillance (AS) case series, the cancer-specific survival for the cohort is 99.7% (Cooperberg et al, 2011), though median follow-up remains relatively short (2–7 years). Active surveillance has emerged as a viable management option and should be offered to patients with low-risk cancer (National Institute for Health and Clinical Excellence, 2008; National Institute of Health, 2011).
There are many different inclusion criteria for AS published in the literature. While all are variations on the model developed by Epstein et al (1994), the discrepancies between them reflect the uncertainty in appropriate cutoffs to distinguish indolent from high-risk cancer. In addition, regional differences in the underlying prevalence of PSA testing in the community alters the pre-test probability for high-risk disease (Moore et al, 2009) in defined ‘low-risk’ cohorts, which potentially interferes with the performance of selection rules when applied to populations distinct from which they were generated.
In our study, we examine application of AS selection rules to a combined Australian, British and Canadian group of patients who underwent radical prostatectomy (RP). Our primary objective was to document the proportion of pathological reclassification from prostate biopsy to RP. Secondary aims included analysis for predictors of high-risk disease and creation of a predictive model to assist with selection of men suitable for AS.
Materials and Methods
Pooled prospectively collected data from Addenbrooke’s Hospital, Cambridge, United Kingdom (2005–2010); The Vancouver Prostate Centre, Canada (1995–2010); and the Australian Prostate Cancer Centre at Epworth, Melbourne, Australia (2003–2010) were retrospectively analysed. All patients had their RP specimens discussed at centralised multidisciplinary meetings and evaluated by dedicated genitourinary pathologists. Cambridge and Vancouver had routine centralised review of biopsies. Ethics approval in all three centres covers the use of collected clinical information for prognostic studies.
A summary of the literature review for published inclusion criteria used for AS is shown in Table 1 (Hardie et al, 2005; Warlick et al, 2006; Dall’Era et al, 2008; Klotz et al, 2009; van den Bergh et al, 2009; Soloway et al, 2010; Adamy et al, 2011). In selecting which AS criteria to apply to our series, we used the University of Toronto criteria, described by Klotz et al (2009), from the first protocol-based AS prospective study and those published from the Prostate Cancer Research International: Active Surveillance (PRIAS) originating from the ERSPC (van den Bergh et al, 2007). Our cohort did not contain data for the amount of cancer present in a biopsy core (length or percentage) as only Cambridge consistently recorded this in their database so the PRIAS criteria were the strictest applicable to our data set.
Patients treated by means of RP who had preoperative parameters appropriate for inclusion for AS per these criteria had final pathology analysed for reclassification rates of upstaging, defined as ⩾pT3, or upgrading, defined as Gleason sum 7–10. Gleason 7 disease was subdivided into 3+4 and 4+3 groups. High-risk disease was defined as ⩾pT3 and/or Gleason sum ⩾8. CAPRA-S scores, a validated postsurgical score to predict PCa recurrence using pre-treatment and pathological data, for risk stratification were also calculated (Cooperberg et al, 2011). Lymph node involvement was not analysed, as across all centres there was no consistent policy regarding the performance of a pelvic lymph node dissection in patients with low-risk disease, and hence data collection was poor.
Differences between groups of continuous variables were determined by Mann–Whitney U or Kruskal–Wallis ANOVA. Pearson’s χ2 or Fisher’s exact test was used to determine differences between groups of categorical variables. To identify predictors of high-risk disease in patients selected for AS, logistic regression models were fitted, including parameters age, PSA, PSAD, clinical stage, number of biopsy cores taken, number of positive biopsy cores and centre of treatment as individual terms. Statistical analyses were performed using SPSS version 18.0 (IBM Corporation, Armonk, NY, USA), and all tests were two-sided with significance assumed at P<0.05 unless otherwise stated.
To generate a clinically usable tool that predicted the presence of high-risk disease in the Cambridge cohort that met the Toronto criteria, all factors found to be significant in multivariate analysis were considered and the most parsimonious model generated from these. Patients were randomly assigned 70:30 to a learning and evaluation cohort. Logistic regression analysis was then performed using all available potential predictors of high-risk disease in the learning cohort, and a nomogram of the resulting equation generated using Orange (http://orange.biolab.si V2.0b, accessed 5 August 2011). The discriminative ability of our nomogram to predict high-risk disease was characterised by generating receiver operating characteristic curves based on the predicted probabilities of the evaluation cohort. A Loess calibration plot was used to assess the performance of our model across the entire range of predicted values, as the tool may have excellent overall accuracy but may not perform well in a specific range of predicted probabilities. A two-component (calibration and AUC calculation) decomposition Brier score was calculated, with a lower Brier score indicating better discriminant properties (Eng, 2006).
Results
Of the 2329 patients that had RP, 800 patients met the Toronto criteria for AS, and this number was reduced to 410 patients when the stricter PRIAS criteria were applied. The preoperative characteristics for these two groups are shown, overall and by treatment centre, in Tables 2a and 2b. The pathology results from RP, including proportions reclassified and final risk group, also divided by treatment centre, are shown in Tables 3a and b. Overall, for those satisfying the Toronto criteria, 50.6% were upgraded to GS ⩾7 and 17.6% upstaged to pT3/4. The reclassification rates for the PRIAS group were 40.5% and 12.4%, respectively. For both groups, the majority of GS upgrading consisted of 3+4 disease (Toronto=84% and PRIAS=79%). In Cambridge, there was a relatively high rate of pT3a disease in the Toronto criteria group (26%) that decreased with PRIAS criteria (14%). Melbourne had a relatively high rate of Gleason 7 disease in final pathology for both the Toronto and PRIAS criteria groups (61 and 52%).
Given that study periods at different centres were different, to account for changes in biopsy technique, pathological interpretation and treatment patterns with time, we repeated the analysis in a restricted cohort (year ⩾2003 and total number of cores taken at biopsy ⩾8) more reflective of contemporary practice. Results of this subanalysis for the Toronto and PRIAS inclusion criteria are shown in Table 4 and were similar to the initial overall analysis with similar proportions of GS upgrading (49.4% Toronto, 41% PRIAS) and pT3/4 upstaging (Toronto 17.9% and PRIAS 11.3%).
Using standard reported clinico-pathological variables, we were unable to derive a more accurate model that predicted indolent disease than the PRIAS criteria. However, we were able to identify significant predictors for high-risk disease (Table 5a) by multivariate logistic regression analysis. For patients meeting the Toronto criteria, increasing age, number of positive cores, the presence of palpable disease as well as the centre of diagnosis were all significant predictors of the presence of high-risk disease; whereas for patients meeting the more stringent PRIAS criteria, only increasing PSA and the presence of palpable disease were significant. Total number of cores taken was analysed and not predictive.
A number of subgroup analyses were undertaken to search for predictors of more advanced disease. Examining for cancer present in a biopsy core (percentage) as a predictor in only the Cambridge cohort did not yield a significant result. Repeating multivariate logistic regression analyses, limited by year of surgery (⩾2003) and number of biopsy cores taken (⩾8) for high-risk disease (⩾pT3 or Gleason sum ⩾8) or ⩾pT3 alone, demonstrated similar predictors to those found for the entire cohort. There were no significant predictors of primary Gleason pattern 4 (as opposed to Gleason sum 8) identified.
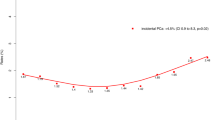
We were unable to generate a nomogram from the whole cohort data because of difficulties modelling sampling error. To account for sampling error, we used an index of prostate size to number of cores taken; however, this did not significantly improve the performance of the model. As the rate of pT3a was high in the Cambridge data, we performed logistic regression analysis to identify predictors of ⩾pT3a/GS⩾8 disease specifically for the Cambridge cohort alone (Table 5b). When only Cambridge data were analysed for the nomogram, PSAD was more predictive than PSA. From this analysis, a nomogram (Figure 1a) was generated that predicts individual risk of high-risk disease in UK men who meet the Toronto criteria, based on age, PSA density, number of positive biopsy cores, and the presence or absence of palpable disease. The logistic regression equation for the nomogram is also included to facilitate future validation studies (Figure 1b).

Nomogram to predict individual probability of high-risk PCa in UK men who meet the Klotz criteria. For a patient, each criteria (age, PSAD, number of positive cores and cT stage) translates to a number of points – read off the top line. The total number of points then corresponds to a probability of finding high-risk PCa if RP is performed. Logistic regression equation for nomogram. Log(p/1-p)=−7.051+0.059(age in years)+0.537(PSAD in units of 0.1 ng ml cm3)+0.996(cT)+0.218(number of positive cores).
Assessment of the nomogram to predict the presence of high-risk disease in a randomly selected evaluation cohort revealed reasonable accuracy (AUC of 0.72) (Figure 2). A calibration plot, comparing nomogram-predicted probabilities to actual proportions of high-risk disease, is shown in Figure 3. This shows that our nomogram underestimates the observed probability of high-risk disease. The two-component (calibration and AUC calculation) decomposition Brier score was 0.199. External validation for the nomogram generated from the Cambridge data, using Melbourne and Vancouver data, showed an AUC of 0.68 and 0.55, respectively.

Receiver operating characteristic curve to assess performance of nomogram predicting probability of high-risk PCa in UK men satisfying Klotz AS criteria.

Calibration plot comparing nomogram-predicted probabilities to actual proportions of high-risk disease.
Discussion
It is clear that published inclusion criteria for AS (Table 1) vary in their stringency. The criteria at John Hopkins Medical Institute are the strictest but some centres elsewhere have accepted Gleason 7 (usually 3+4), PSA levels up to 15 ng ml−1 and all clinical T2 disease. Furthermore, not all centres use PSAD (⩽0.15–0.20), number of positive cores (⩽2, ⩽3 or 1/3 of total) and percentage (⩽20–50%) of single core involvement to enrol patients. It has been demonstrated previously (Suardi et al, 2008; Conti et al, 2009; Mufarrij et al, 2010) that increases in stringency can decrease the rates of adverse pathological features but will also substantially decrease the number of men suitable for AS. Until the ProtecT trial reports (Lane et al, 2010), we will not know the criteria that predict the most important outcome of AS, namely death from PCa.
Our paper reports combined results from the Australian, British and Canadian academic centres. These results are compared with the American (UCSF, n=331 (Conti et al, 2009)) and European (Milan and Hamburg, n=2455 and Milan, n=85 (Suardi et al, 2008; Suardi et al, 2010)) cohorts, where the Toronto and PRIAS inclusion criteria were also used to select patients from a RP database. Applying the Toronto criteria, we found a higher than previously reported rate of Gleason score upgrading (50.6%) compared with Conti et al 31% and Suardi et al 38.1%. However, similar to previous reports, the majority of GS upgrading was 3+4 disease (84%) and this may not translate into a major clinical problem. The rate of upstaging (EPE/SVI) when using the Toronto criteria (17%) was similar to previous reports in the literature (14–15%). There was less upstaging and upgrading when using the stricter PRIAS criteria; however, our reclassification rates are markedly higher (upgrading 42.7% and upstaging 12.7%) compared with those previously reported by the Milan group (upgrading 7.1% and upstaging 2.4%; Suardi et al, 2008). The results of our subanalysis, by year (⩽2003) and number of biopsy cores taken (⩾8), demonstrated similar predictors to those found for the entire cohort. This was not surprising given that the two cohorts with high rates of upstaging (Cambridge) and upgrading (Melbourne) were the most contemporary with higher median number of cores taken (Tables 2a and b).
Individually, each of our three centres has a relatively high rate of GS upgrading (Table 4, Cambridge 44%, Vancouver 47%, Melbourne 62%). Possible reasons for this include sampling error on biopsy, interobserver pathology variation between biopsy and RP reports and differing geographic population patterns of disease. The number of biopsy cores taken for each centre was: Cambridge (median=12, IQR10-12), Melbourne (median=10, IQR8-13) and Vancouver (median=8, IQR8-8). The standard extended core template for prostate biopsy consists of 10–12 cores, and hence particularly in Vancouver biopsy under-sampling could affect our results. To minimise biopsy sampling error, Adamy et al (2011) suggest immediate confirmatory biopsy before commencing AS. There may be inter-observer variation influencing the Melbourne data as ∼50% of biopsies are reported by community pathologists, whereas all RP specimens are read by two specialist uro-pathologists. In addition, most of the upgrading was predominantly Gleason 3+4 disease (84% of those upgraded (Suardi et al, 2008)). Results for carefully selected men on AS with intermediate-risk disease (Gleason sum 7 or CAPRA score 3–5) have been published suggesting that within limited follow-up (4years) outcomes were similar to men with GS 3+3 disease (Cooperberg et al, 2011).
Cambridge had a high proportion of EPE in its Toronto criteria group (26% vs 12%, P<0.001) compared with the other two centres. As pathology for biopsy and RP is centrally reviewed in Cambridge and an extended template used for biopsy, this high rate of pT3a disease could also be attributable to a low uptake of PSA testing in the United Kingdom. Melia (2005) compared worldwide rates of PSA testing in 2005 and, allowing for lack of standardized data, found rates of PSA testing in the United Kingdom considerably lower than elsewhere. At a similar period of time (year 2000), the rate of PSA testing in the United Kingdom was 5.4 tests per 100 men per annum (men 45–84 years old) compared with the United States, 38% of black men and 31% of white men (⩾65 years); Italy, 26.9% men (⩾40 years); Australia, 23% men (40–70 years); and Canada (Beaulac et al, 2006), 47.5% (⩾50 years).
The few previously published predictive tools for AS selection have focused on calculating likelihood of indolent, low-volume/low-grade or insignificant PCa rather than high-risk features (Kattan et al, 2003; Ochiai et al, 2005; Nakanishi et al, 2007; Steyerberg et al, 2007; O’Brien et al, 2011). Nakanishi et al’s nomogram, using a cohort of 258 men from the Canada and the United States, is specific for men with a single-positive biopsy core and uses age, PSAD and tumour length in a core to predict indolent cancer (Nakanishi et al, 2007). Possibly having a single positive core is too stringent a criterion for a programme of AS. Kattan et al ‘s nomogram to predict small, moderately differentiated, confined tumours (Kattan et al, 2003) that was recently validated and updated (Steyerberg et al, 2007); however, its application is questionable as its data are based on sextant biopsies with most centres now performing 10–12 core biopsies. A nomogram generated from an Australian cohort was recently published (O’Brien et al, 2011), and for multiple probability cutoffs predicting indolent PCa, they gave coexisting rates of high-risk disease. It is likely that in the future men will be selected for AS programmes only if they have an mp-MRI that does not demonstrate any other unsuspected cancers, possibly coupled with a limited template biopsy. The result of the PIVOT trial (Wilt et al, 2012) has demonstrated that for low-risk men, surgery offers limited advantages over watchful waiting, and AS programmes are likely to be at least as effective.
To our knowledge, there has not been a nomogram derived from the British data to assist with selection of patients for AS. From our Cambridge data, we generated a nomogram that predicts presence of high-risk disease in patients who satisfy the Toronto entry criteria for AS. Overall assessment of the performance of our nomogram was good (AUC 0.72 and two-component Brier score 0.199). However, the calibration plot suggests that our nomogram consistently underestimates the observed proportion of high-risk disease for nearly all predicted values. Unsurprisingly, the nomogram performed poorer when we used the Melbourne (AUC 0.68) and Vancouver (0.55) data to externally validate it. There is evidence that risk calculators are best applied to the population from which they are generated (Bhojani et al, 2009). An ideal nomogram would also include information on previous biopsies, family history of significant CaP and results of MRI imaging; however, this was not present in our data set. Given the low rate of PSA testing in the United Kingdom and the high rate of upstaging in our Cambridge cohort, our nomogram would be a reasonable tool for counselling UK patients in regards to AS.
Our study has limitations. Being retrospective, data collected were reliant on individual centres’ protocols and they did not include all information on tumour volume. The use of additional criteria such as length or percentage of a single core involved might reduce the amount of reclassification, but would likely reduce the number of patients to whom AS could be offered. Data on lymph node status were lacking as lymph node dissection for low-risk disease was according to surgeon preference, and not consistently performed or recorded. Lack of follow-up data for cancer recurrence or death is significant, as having pathological features of advanced disease on biopsy does not necessarily translate to poorer outcomes after surgery. Using a surgical cohort includes unforeseen biases in patient selection not addressed in AS criteria, such as age, comorbidities, family history, patient anxiety for intervention, and findings on imaging or institutional bias towards type of treatment. The median age of our cohort (61 years) is younger than that reported by AS cohorts (65 years; Carter et al, 2007; van den Bergh et al, 2009). Being a multicentre study, multiple pathologists reported biopsies and specimens, and the effect of inter-observer variation was not calculated. We also accept that, given that our study spanned a broad period of time, interpretive changes in pathological grading and clinical staging of PCa did occur (Thompson et al, 2005). Multiple surgeons performed the surgery though positive margin rates that were similar.
Conclusions
Our study examined the rates of reclassification of men from Australia, Britain and Canada who underwent RP who initially would have been suitable for AS, as defined by the Toronto and PRIAS criteria. Compared with previously reported cohorts from Europe and North America using the same AS selection criteria, we found significantly higher rates of upgrading and upstaging.
Care must be used when applying AS criteria generated from one population to another distinct population. There is an onus on larger centres in individual countries to assess the performance of different criteria on their population before implementation in routine AS programmes and generate predictive tools from their own data sets. Use of increasingly stringent selection criteria may reduce reclassification but this must be balanced against the exclusion of a significant number of men from AS who may benefit from such an approach.
The acceptability of AS protocols would best be evaluated by ongoing prospective studies. The development of novel serum or tissue markers, and improved imaging to predict disease progression would help remove any uncertainties physicians and patients have with AS.
Change history
11 October 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adamy A, Yee DS, Matsushita K, Maschino A, Cronin A, Vickers A, Guillonneau B, Scardino PT, Eastham JA (2011) Role of prostate specific antigen and immediate confirmatory biopsy in predicting progression during active surveillance for low risk prostate cancer. J Urol 185 (2): 477–482
Beaulac JA, Fry RN, Onysko J (2006) Lifetime and recent prostate specific antigen (PSA) screening of men for prostate cancer in Canada. Can J Public Health 97 (3): 171–176
Bhojani N, Salomon L, Capitanio U, Suardi N, Shahrokh FS, Jeldres C, Zini L, Pharand D, Ois Pe Loquin O, Arjane P, Abbou C, De La Taille A, Montorsi F, Karakiewicz P (2009) External validation of the updated Partin tables in a Cohort of French and Italian men. Int J Radiat Oncol Biol Phys 73 (2): 347–352
Carter HB, Kettermann A, Warlick C, Metter EJ, Landis P, Walsh PC (2007) Expectant management of prostate cancer with curative intent: an update of the Johns Hopkins experience. J Urol 178: 2359
Conti SL, Dall’Era M, Fradet V, Cowan JE, Simko J, Carroll PR (2009) Pathological outcomes of candidates for active surveillance of prostate cancer. J Urol 181: 1628–1634
Cooperberg MR, Cowan JE, Hilton JF, Reese AC, Zaid H, Porten SP, Shinohara K, Meng VM, Greene KL, Carroll PR (2011) Outcomes of active surveillance for men with intermediate-risk prostate cancer. JCO 29 (2): 228–234
Cooperberg MR, Hilton JF, Carroll PR (2011) The CAPRA-S score: a straightforward tool for improved prediction of outcomes after radical prostatectomy. Cancer 117: 5039–5046
Dall’Era MA, Konety BR, Cowan JE, Shinohara K, Stauf F, Cooperberg MR, Meng VM, Kane CJ, Perez N, Master VA, Carroll PR (2008) Active surveillance for the management of prostate cancer in a contemporary cohort. Cancer 112: 2664
Eng J (2006) (Updated 17 May 2006 (accessed 14 February 2011)) ROC Analysis: Web-based Calculator for ROC Curves. Johns Hopkins University: : Baltimore., Available from: http://www.jrocfit.org
Epstein JI, Walsh PC, Carmichael M, Brendler CB (1994) Pathologic and clinical findings to predict tumor extent of nonpalpable (stage T1c) prostate cancer. JAMA 271: 368
Hardie C, Parker C, Norman A, Eeles R, Horwich A, Huddart R, Dearnaley D (2005) Early outcomes of active surveillance for localized prostate cancer. BJU Int 95: 956–960
Kattan MW, Eastham JA, Wheeler TM, Maru N, Scardino PT, Erbersdobler A, Graefen M, Huland H, Koh H, Shariat SF, Slawin KM, Ohori M (2003) Counseling men with prostate cancer: a nomogram for predicting the presence of small, moderately differentiated, confined tumors. J Urol 170: 1792–1797
Klotz L, Zhang L, Lam A, Nam R, Mamedov A, Loblaw A (2009) Clinical results of long-term follow-up of a large, active surveillance cohort with localized prostate cancer. J Clin Oncol 28: 126–131
Lane JA, Hamdy FC, Martin RM, Turner EL, Neal DE, Donovan JL (2010) Latest results from the UK trials evaluating prostate cancer screening and treatment: the CAP and ProtecT studies. Eur J Cancer 46 (17): 3095–3101
Lin K, Croswell JM, Koenig H, Lam C, Maltz A (2011) Prostate-Specific Antigen-Based Screening for Prostate Cancer: An Evidence Update for the U.S. Preventive Services Task Force [Internet]. Evidence syntheses, No. 90, Agency for Healthcare Research and Quality: Rockville, MD, Available from: http://www.ncbi.nlm.nih.gov/books/NBK82303/
Melia J (2005) Part 1: The burden of prostate cancer, its natural history, information on the outcome of screening and estimates of ad hoc screening with particular reference to England and Wales. BJU Int 95 (Suppl 3): 4–15
Moore AL, Dimitropoulou P, Lane A, Powell PH, Greenberg DC, Brown CH, Donovan JL, Hamdy FC, Martin RM, Neal DE (2009) Population-based prostate-specific antigen testing in the UK leads to a stage migration of prostate cancer. BJU Int 104 (11): 1592–1598
Mufarrij P, Sankin A, Godoy G, Lepor H (2010) Pathologic outcomes of candidates for active surveillance undergoing radical prostatectomy. Urology 76 (3): 689–692
Nakanishi H, Wang X, Ochiai A, Trpkov K, Yilmaz A, Donnelly JB, Davis JW, Troncos P, Babaian RJ (2007) A nomogram for predicting low-volume/low-grade prostate cancer. A tool for selecting patients for active surveillance. Cancer 110 (11): 2441–2447
National Institute of Health (2011) Role of active surveillance in the management of men with localized prostate cancer. Draft Statement. National Institutes of Health State-of-the-Science Conference: Role of Active Surveillance in the Management of Men With Localized Prostate Cancer, 5–7 December 2011.
National Institute for Health and Clinical Excellence (2008) NICE Prostate Cancer: Diagnosis and Treatment. A Systematic Review (cited 13 May 2009). Available from http://www.nice.org.uk/nicemedia/pdf/CG58NICEGuideline.pdf
O’Brien BA, Cohen RJ, Ryan A, Sengupta S, Mills J (2011) A new preoperative nomogram to predict minimal prostate cancer: accuracy and error rates compared to other tools to select patients for active surveillance. J Urol 186: 1811–1817
Ochiai A, Troncoso P, Chen ME, Lloreta J, Babaian RJ (2005) The relationship between tumor volume and the number of positive cores in men undergoing multisite extended biopsy: implication for expectant management. J Urol 174: 2164–2168
Soloway MS, Soloway CT, Eldefrawy A, Acosta K, Kava B, Manoharan M (2010) Careful selection and close monitoring of low-risk prostate cancer patients on active surveillance minimizes the need for treatment. Eur Urol 58: 831–835
Steyerberg EW, Roobol MJ, Kattan TH, van der Kwast TH, Koning HJ, Schroder FH (2007) Prediction of indolent prostate cancer: validation and updating of a prognostic nomogram. J Urol 177: 107–112
Suardi N, Briganti A, Gallina A, Salonia A, Karakiewicz PI, Capitanio U, Freschi M, Cestari A, Guazzoni G, Rigatti P, Montorsi F (2010) Testing the most stringent criteria for selection of candidates for active surveillance in patients with low-risk prostate cancer. BJU Int 105 (11)): 1548–1552
Suardi N, Capitanio U, Chun FKH, Graefen M, Perrotte P, Schlomm T, Haese A, Huland H, Erbersdobler A, Montorsi F, Karakiewicz PI (2008) Currently used criteria for active surveillance in men with low-risk prostate cancer. An analysis of pathologic features. Cancer 113 (8): 2068–2072
Thompson IM, Canby-Hagino E, Lucia MS (2005) Stage migration and grade inflation in prostate cancer: Will Rogers meets Garrison Keillor. J Natl Cancer Inst 97: 1236–1237
van den Bergh RCN, Roemeling S, Roobol MJ, Roobol W, Schroder FH, Bangma CH (2007) Prospective validation of active surveillance in prostate cancer: the PRIAS study. Eur Urol 52: 1560–1563
van den Bergh RC, Roemeling S, Roobol MJ, Aus G, Hugosson J, Rannikko AS, Tammela TL, Bangma CH, Schroder FH (2009) Outcomes of men with screen-detected prostate cancer eligible for active surveillance who were managed expectantly. Eur Urol 55: 1–8
Warlick C, Trock BJ, Landis P, Epstein JI, Carter HB (2006) Delayed versus immediate surgical intervention and prostate cancer outcome. J Natl Cancer Inst 98: 355–357
Wilt TJ, Brawer MK, Jones KM, Barry MJ, Aronson WJ, Fox S, Gingrich JR, Wei JT, Gilhooly P, Grob BM, Nsouli I, Iyer P, Cartagena R, Snider G, Roehrborn C, Sharifi R, Blank W, Pandya P, Andriole GL, Culkin D . Wheeler for the Prostate Cancer Intervention versus Observation Trial (PIVOT) Study Group (2012) Radical prostatectomy versus observation for localized prostate cancer. NEJM 367 (3): 203–213
Acknowledgements
We would like to thank Dr Karan Wadhwa for assistance with data entry in Cambridge.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wong, LM., Neal, D., Johnston, R. et al. International multicentre study examining selection criteria for active surveillance in men undergoing radical prostatectomy. Br J Cancer 107, 1467–1473 (2012). https://doi.org/10.1038/bjc.2012.400
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.400
Keywords
This article is cited by
-
Old men with prostate cancer have higher risk of Gleason score upgrading and pathological upstaging after initial diagnosis: a systematic review and meta-analysis
World Journal of Surgical Oncology (2021)
-
Predictors of adverse pathology on radical prostatectomy specimen in men initially enrolled in active surveillance for low-risk prostate cancer
World Journal of Urology (2021)
-
Establishment and Validation of Extra-transitional Zone Prostate Specific Antigen Density (ETzD), a Novel Structure-based Parameter for Quantifying the Oncological Hazard of Prostates with Enlarged Stroma
Scientific Reports (2019)
-
Diagnostic prostate biopsy performed in a non-academic center increases the risk of re-classification at confirmatory biopsy for men considering active surveillance for prostate cancer
Prostate Cancer and Prostatic Diseases (2015)
-
Evaluation of models predicting insignificant prostate cancer to select men for active surveillance of prostate cancer
Prostate Cancer and Prostatic Diseases (2015)
