
Abstract
Background:
Docetaxel is widely used as a chemotherapeutic agent for gastric cancer treatment. A combined regimen with sunitinib demonstrated a synergistic antitumour effect in a preclinical model. The aim of this study was to evaluate the efficacy and safety of this combination in patients with unresectable or metastatic advanced gastric cancer following failure of treatment with a fluoropyrimidine and platinum combination.
Methods:
This open-label, phase II, randomised trial enrolled patients with unresectable or metastatic gastric cancer. Patients were assigned to either a docetaxel monotherapy arm (D only arm: 60 mg m−2, every 3 weeks) or a combination arm (DS arm: docetaxel+sunitinib 37.5 mg every day). The primary end point of the study was time to progression and the secondary end points were overall response rate, disease control rate, overall survival, and toxicity profile. A pharmacokinetic study was also performed.
Results:
A total of 107 patients were entered into the study. The TTP was not significantly prolonged in the DS arm when compared with the D only arm (DS vs D only arm: 3.9 months (95% confidence interval (CI) 2.9–4.9) vs 2.6 months (95% CI 1.8–3.5) (P=0.206). The hazard ratio for TTP was 0.77 (95% CI 0.52–1.16). However, the objective response rate was significantly higher in the DS arm (41.1% vs 14.3%, P=0.002). Patients in the DS arm experienced stomatitis, diarrhoea, and hand–foot syndrome more frequently.
Conclusion:
The addition of sunitinib to docetaxel did not significantly prolong TTP, although it significantly increased response.
Similar content being viewed by others


Main
Gastric cancer is the fourth most common cancer worldwide, with nearly one million new cases diagnosed every year (Jemal et al, 2011). It is also one of the leading causes of cancer-related mortality in Asia (Jung et al, 2011; Tanaka et al, 2011). For patients with recurrent or metastatic disease, chemotherapy can improve survival and can also possibly provide significant palliation of symptoms (Pyrhonen et al, 1995; Glimelius et al, 1997). In terms of first-line regimens, a combination of fluoropyrimidine and platinum is regarded as a standard option as this combination has shown superior clinical outcomes in phase III trials when compared with fluoropyrimidine monotherapy and other combinations (Wagner et al, 2006). However, less than half of the patients achieve an objective response and the duration, even in these responders, is as short as a few months (Van Cutsem et al, 2002).
Recently, we have conducted a randomised phase III trial to compare best supportive care vs best supportive care plus chemotherapy (irinotecan or docetaxel) following the failure of fluropyrimidine/platinum-based chemotherapy for the treatment of gastric cancer. In this phase III trial, 193 gastric cancer patients were randomly assigned to second-line chemotherapy (n=128) or best supportive care (n=65). A significant prolongation in survival was observed for the second-line chemotherapy arm when compared with best supportive care (hazard ratio, 0.63; 95% CI, 0.47–0.86; P=0.004) (Park et al, 2011). Thus, second-line chemotherapy should be considered in selected patients with gastric cancer.
Docetaxel is one of the most widely used chemotherapeutic agents for the second-line treatment of gastric cancer. Docetaxel exerts its antitumour activity by stabilising microtubules. It also inhibits the anti-apoptotic gene Bcl2 and promotes the expression of p27, a cell-cycle inhibitor, as well as other pro-angiogenic factors, such as vascular endothelial growth factor (VEGF) (Nishiyama and Wada, 2009). As a monotherapy, this agent has shown an overall response rate of around 15% and time to progression (TTP) of 2.5–3 months (Bang et al, 2002; Giuliani et al, 2003; Lee et al, 2008). Combination of docetaxel with other agents, such as platinum as a treatment for gastric cancer, has failed to demonstrate any significant improvement in efficacy in most studies, when compared with docetaxel alone (Park et al, 2004; Nguyen et al, 2006; Barone et al, 2007; Zhong et al, 2008; Kim et al, 2011). Although one study has shown that the addition of docetaxel was associated with improved clinical outcomes (Van Cutsem et al, 2006), docetaxel is not widely recommended as a standard treatment option because of its high rate of toxicity, including neutropenia and neurotoxicity, and also the need for subsequent discontinuation of the treatment (Ilson, 2007).
Sunitinib is an oral inhibitor of multiple receptor tyrosine kinases that are involved in tumour proliferation and angiogenesis, specifically platelet-derived growth factor receptor, VEGF receptor, KIT, Flt-3, and RET (Abrams et al, 2003a; Mendel et al, 2003; O’Farrell et al, 2003). This agent has shown efficacy against many solid cancers, including metastatic clear cell renal cell carcinoma (Motzer et al, 2009), imatinib-refractory gastrointestinal stromal tumour (Demetri et al, 2006), and pancreatic neuroendocrine tumour (Raymond et al, 2011). In preclinical studies, docetaxel and sunitinib demonstrated additive antitumour activity in mouse xenograft models of non-small-cell lung cancer and breast cancer (Abrams et al, 2003b; Christensen JG). Recently, in a multicentre, phase II trial, single-agent sunitinib given in a second-line setting demonstrated an overall response rate of 2.6% with 32.1% of stable disease lasting more than 6 weeks and manageable toxicity in patients with advanced gastric cancer (AGC) (Bang et al, 2010). In the present study, we conducted a randomised, phase II trial of docetaxel singly or in combination with sunitinib in patients who had experienced failure of fluoropyrimidine and platinum therapy. This study is registered at clinicaltrials.gov as #NCT01238055.
Materials and methods
Study design
This was an open-label, phase II, two arm, randomised, single centre study conducted at Samsung Medical Center, Seoul, Korea. The protocol was approved by the institutional review board at Samsung Medical Center and the trial was conducted in accordance with the Declaration of Helsinki. All patients were required to give written informed consent before enrolment. Pfizer (Seoul, Korea) provided sunitinib gratis, but was not involved in the accrual or analysis of the data or in the preparation of the manuscript.
Patients
Patients aged ⩾18 years with unresectable or metastatic adenocarcinoma of the stomach or gastroesophageal junction, whose cancer had progressed after a first-line fluoropyrimidine and platinum combination regimen, were eligible to enter the study. Other eligibility criteria were as follows: measurable or evaluable disease according to the Response Evaluation Criteria in Solid Tumours (RECIST) 1.1; performance status 0–2 by Eastern Cooperative Oncology Group scale; adequate organ function including bone marrow (absolute neutrophil count ⩾1500 μl−1, platelet ⩾100 000 μl−1), liver (AST/ALT⩽2.5 × ULN, 5.0 × ULN if liver involvement, total serum bilirubin ⩽2.0 mg dl−1) and kidney (serum creatinine ⩽1.5 × ULN); life expectancy of more than 3 months; and written informed consent. Patients with severe co-morbid illness and/or active infections, grade 3 or higher haemorrhage according to CTCAE v3.0 (US National Cancer Institute) within prior 4 weeks, pregnant or lactating women, or active CNS metastases not controllable with radiotherapy or corticosteroids were excluded from the study.
Treatment
Patients were randomised in 1 : 1 ratio either to a docetaxel plus sunitinib (DS arm) arm or to a docetaxel single arm (D only arm) using a random permuted block design. Random assignment was stratified by ECOG performance status (0, 1 vs 2). Docetaxel was administered at the dose of 60 mg m−2 on day 1 of each 3-week cycle and sunitinib was administered orally at the starting dose of 37.5 mg daily, on a continuous daily dosing schedule. Tumour evaluation was performed every two cycles and response to chemotherapy was assessed in accordance with the RECIST criteria v1.1. Clinical laboratory evaluations (biochemistry, haematology, and urinalysis) were carried out on day 1 of every cycle. Toxicity assessment adhered to CTCAE v3.0. Treatment was discontinued in case of tumour progression, unacceptable toxicity, or consent withdrawal. No crossover to the DS arm was allowed for patients in the D only arm after progression.
For patients receiving docetaxel administration, if grade 4 neutropenia, febrile neutropenia, skin/nail changes, or grade 2 peripheral neuropathy developed, dose was reduced to 45 mg m−2. If a patient continued to experience these toxicities (other than neuropathy) at 45 mg m−2, treatment was discontinued. In terms of peripheral neuropathy, treatment was discontinued if grade 3 or 4 toxicity was developed. For patients receiving sunitinib administration, if grade 3 toxicity developed, sunitinib was withheld until the toxicity was resolved to <grade 2, then treatment was reduced to 25 mg. If a second occurrence of >grade 1 toxicity occurred, sunitinib was further reduced to 12.5 mg. At a third occurrence of >grade 1 toxicity, sunitinib was discontinued.
Pharmacokinetic analysis
Average steady-state plasma concentrations (Css,av) for sunitinib and its active metabolite SU12662 were measured on the last day of cycles 2, 4, and 6, before drug administration of the next cycle. All the samples were centrifuged at 2092 g for 10 min immediately after collection, and stored frozen at −70 °C until assayed. Plasma concentrations of sunitinib and SU12662 were determined by high performance liquid chromatography coupled with tandem mass spectrometry. The lower limit of quantification was 0.25 ngml−1 for sunitinib and SU12662.
Data analysis was performed on the intent-to-treat population using the last observation carried forward method for missing data. Dose-normalised Css,av of sunitinib and SU12662 at each cycle was analysed by repeated-measures ANOVA (SAS, version 9.1; SAS Institute Inc., Cary, NC, USA).
Statistics
The primary end point of the current study was TTP, which was defined as the interval between the date of first study treatment and the date of documented disease progression. The secondary end points were objective response rate (ORR), disease control rate (DCR), and overall survival (OS), which was calculated from the date of first study treatment to the date of death and toxicity profile. TTP and OS were calculated by the Kaplan–Meier product–limit method.
For sample size calculation, we initially assumed 12 months of accrual period and 6 months of follow-up period. The current study was designed with two-sided, α=0.05, 90% power to detect a null median TTP of 2 months and experimental median TTP of 4 months (N=108). Assuming a 10% dropout rate, total target accrual number was 116 (108+10%). The sample size was recalculated on February 2011 owing to a longer patient accrual period (>24 months) than originally planned (12 months). The revised sample size calculation was designed with two-sided, α=0.05, 90% power to detect a null median TTP of 2 months and experimental median TTP of 4 months (N=108) after a 36-month accrual period and 18-month follow-up (N=92). We amended the protocol to accrue 102 patients. The laboratory data and prognostic factors obtained from demographic data on the baseline were evaluated by Pearson’s Chi-square test for patient homogeneity on the baseline between the two arms and the associations among the prognostic factors. The efficacy and toxicity of the two arms were evaluated by comparing response rate, DCR, and incidence of toxicity using Pearson’s chi-square tests, while TTP and OS were compared by log-rank tests. For pharmacokinetic study, the protocol was amended (at the time of 80 patient accrual) to enrol patients for pharmacokinetic blood sampling. Those patients who had consented for pharmacokinetic sampling, which was not mandatory, were enrolled.
Results
Patient characteristics
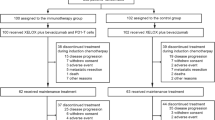
Between December 2008 and February 2011, 107 patients were entered into the study. As 2 patients withdrew their consent before treatment, clinical outcomes and toxicities were evaluated in 105 patients. The median age of the patients was 53 years (20–72) and male : female ratio was 73 (69.5%) : 32 (30.5%). The ECOG performance scale indicated a grade of 0–1 for 96 (91.4%) patients. After randomisation, 56 patients (53.3%) were allocated to the DS arm and 49 patients (46.7%) to the docetaxel arm (D only arm). Demographics of the two arms, including median age, sex ratio, ECOG performance scale, laboratory findings (except serum alkaline phosphatase level), and sites of metastatic lesions, were not significantly different. These details are provided in Table 1.
Pharmacokinetic analysis
A total of 66 plasma concentrations (33 for sunitinib, 33 for SU12662) from 13 patients were analysed. No differences were noted in the dose-normalised Css,av of sunitinib and SU12662 over the cycle using repeated-measures ANOVA (Table 2 and Figure 1).

Dose-normalised Css,av of sunitinib and SU12662 at each cycle.
Treatment response and survival
A total of 423 cycles were administered, with a median of three cycles given per patient (range 1–18). No significant difference was noted between the DS and D only arms in terms of the median number of treatment cycles (P=0.283). At the time of analysis, 97 cases (92.3%) of progression had occurred, including 52 cases (92.9%) in the DS arm and 45 cases (91.8%) in the D only arm. The TTP was not significantly prolonged in the DS arm when compared with the D only arm (DS vs D only arm: 3.9 months (95% confidence interval (CI) 2.9–4.9) vs 2.6 months (95% CI 1.8–3.5), P=0.206) (Figure 2). The hazard ratio for TTP was 0.77 (95% CI 0.52–1.16). The response rate was significantly higher for the DS arm than for the D only arm (DS vs D only arm: 41.1% (95% CI 28.1–55.0) vs 14.3% (95% CI 6.0–27.2), P=0.002) (Table 3). Notably, one patient with multiple liver metastases achieved complete remission after 4 cycles of docetaxel and sunitinib. The patient is still maintaining complete remission after completion of 4 cycles of docetaxel and sunitinib (that is, for more than 2 years) at the time of this writing (Figure 3). The DS arm had 1 CR, 29 PRs, 37 SDs, and 38 PDs, by RECIST 1.1. All treatment responses were centrally reviewed while blinded to the treatment arm. A similar tendency was observed for the DCR, which was 75.0% (42/56, 95% CI 61.6–85.6) in the DS arm and 51.0% (25/49, 95% CI 36.3–65.6) in the D only arm (P=0.011, Table 3).

Time to progression curve of the patients.

A case of complete response of docetaxel/sunitinib combination. (A) Baseline image. (B) After four cycles of treatment.
The DS regimen did not significantly improve survival when compared with the D only regimen (DS vs D only arm: 8.0 (95% CI 5.4–10.6) vs 6.6 (95% CI 3.6–9.7), P=0.802) (Figure 4). The hazard ratio for OS was 0.94 (95% CI 0.60–1.49).

Overall survival curve of the patients.
Adverse events
Among the 105 patients whose toxicity profile was assessed, 64 patients (61.0%) experienced grade at least one grade 3–4 adverse event (AE). The most common grade 3–4 AE was neutropenia, which occurred in 18 patients (32.1%) in the DS arm and 10 patients (20.4%) in the D only arm. Overall, the incidence of grade 3–4 AEs was not significantly different between the two arms (46.4% (26/56) in the DS arm, 30.6% (15/49) in the D only arm, P=0.112). In addition, no statistical difference was observed between the two groups in terms of haematological toxicity (including anaemia, leukopenia, neutropenia, thrombocytopenia, and febrile neutropenia), nausea, vomiting, neuropathy, and myalgia. However, patients who received docetaxel and sunitinib had a greater chance of experiencing stomatitis, diarrhoea, and hand–foot syndrome (HFS) than did those who received docetaxel only. These details are described in Table 4.
The occurrence of these AEs led to at least one cycle of dose reduction in 46 (43.8%) patients and treatment delay in 57 (54.3%) patients. The DS arm showed 39 and 50 cases of dose reduction and treatment delay, respectively, out of 230 cycles of treatment. The D only arm showed 23 and 44 cases of dose reduction and treatment delay, respectively, out of 193 cycles. Occurrences of dose reduction (29/56, 51.8% in arm A, 17/49, 34.7% in arm B, P=0.078) and treatment delay (32/56, 57.1% in arm A, 25/49, 51% in arm B, P=0.530) were not significantly different between the two groups. The dose intensity was 65% of the planned dose for docetaxel and 60% of the planned dose for sunitinib.
Discussion
In this phase II randomised study, we analysed the efficacy and toxicity of 3-weekly administration of docetaxel 60 mg m−2 combined with daily administration of sunitinib 37.5 mg in patients with AGC who had failed to respond to first-line fluoropyrimidine and platinum. The current study showed the following findings: (1) combination of docetaxel and sunitinib demonstrated a median TTP of 3.9 months (95% CI 2.9–4.9), while docetaxel monotherapy demonstrated a median TTP of 2.6 months (95% CI 1.8–3.5) (P=0.206). Importantly, patients who had received the combined treatment showed significantly higher ORR (DS vs D only arm: 41.1% vs 14.3%, P=0.002). However, patients in the DS arm experienced stomatitis, diarrhoea, and HFS more frequently than did patients in the D only arm.
In the current study, 56 patients received docetaxel combined with sunitinib. Importantly, one patient who had multiple liver metastases from gastric cancer, and who had failed to respond to previous TS-1/cisplatin chemotherapy, achieved complete remission after docetaxel/sunitinib and has maintained remission for >2 years (Figure 3). The high response rate with the docetaxel and sunitinib combination in gastric cancer suggests that this regimen might be feasible in a neoadjuvant setting where response rate is important. Although the DS regimen did not prolong TTP in all GC patients, the regimen substantially increased response rate and thus may be important as a preoperative regimen in gastric cancer patients with limited metastases. In addition, the docetaxel and sunitinib combination may be tested in the first-line setting for gastric cancer. Based on our phase III trial which compared docetaxel, irinotecan and supportive care in salvage setting for gastric cancer, irinotecan monotherapy seemed better than docetaxel alone (Park et al, 2011). However, at the time of the study design, the phase III trial was ongoing and there was lack of preclinical data for irinotecan and sunitinib combination. There are few phase I dose-finding studies for the combination of sunitinib and docetaxel (Robert et al, 2010; de Jonge et al, 2011). The recommended dose for the combination was docetaxel 75 mg m−2 q 3 weeks and sunitinib 37.5 mg daily. Initially, we had planned to administer the above regimen. However, the first two patients had grade 4 neutropenia with febrile episode. After internal discussion with the internal safety monitoring board, we had decided to lower the dose to docetaxel 60 mg m−2 and sunitinib 37.5 mg daily every 3 weeks.
The PK results of sunitinib and SU12662 obtained at steady-state were similar to those from single-agent trials in patients with advanced solid malignancies (Britten et al, 2008). As mentioned earlier, no difference was observed for the dose-normalised Css,av of sunitinib and SU12662 over the cycle. Therefore, the conclusion may be made that autoinduction or autoinhibition of sunitinib or SU12662 metabolism has not been observed with prolonged dosing.
Identification of predictive biomarkers that could identify the subset of gastric cancer patients who might dramatically respond to docetaxel and sunitinib will be clinically important. Although our study failed to demonstrate improved TTP in the docetaxel and sunitinib combination arm, it did indicate that some gastric cancer patients may respond dramatically to a combined docetaxel and sunitinib treatment. Identification of key biomarkers that might predict treatment response to sunitinib will aid clinicians in designing future enriched clinical trials on sunitinib as a treatment for metastatic gastric cancer. Currently, no specific biomarker has been identified that can predict treatment response to sunitinib.
The German Arbeitsgemeinschaft Internistische Onkologie (AIO) study investigated the associations between serum VEGF-C concentrations and treatment outcome in a phase II sunitinib monotherapy trial for gastric cancer (Moehler et al, 2011). However, owing to limited tissue availability, the biomarker study in the AIO study was not conclusive. Recent pharmacogenetic analysis has revealed that patients harbouring certain single-nucleotide polymorphisms (SNPs) are predisposed to experience more frequent toxicities or lower response (van Erp et al, 2009; Garcia-Donas et al, 2011) when treated with sunitinib. We have collected serial blood samples as well as paraffin-embedded tissues for further correlative analyses and soon will report the predictive and prognostic role of those SNPs in sunitinib treatment.
Grade 3–4 neutropenia, which is a serious toxicity of the docetaxel and sunitinib combination, occurred in 18 patients (32.1%) in the DS arm and 10 patients in the D arm (P=0.169). Overall, the incidences of other haematological toxicities including anaemia, leukopenia, or thrombocytopenia were not significantly different between the two groups. The incidence of febrile neutropenia was not significantly different in the two arms (P=0.196) and no subsequent mortality was observed. Although this could be the result of the small number of the recruited patients, we authors do not consider this AE to be a major problem for the docetaxel and sunitinib combination treatment.
However, in the DS arm, the incidences of any grade of stomatitis, diarrhoea, and HFS were more frequent than in the D arm, with statistical significance (any grade stomatitis: 51.8% vs 14.3% (P<0.001); any grade diarrhea: 35.7% vs 14.2% (P=0.033); any grade HFS: 53.6% vs 20.4% (P=0.002)). Because of these AEs, a substantial number of patients needed treatment modifications. Patients who had received the DS treatment showed a tendency to experience more frequent dose reductions, but the difference was not statistically significant (29/56, 51.8% in arm DS, 17/49, 34.7% in arm D, P=0.078). Therefore, we might conclude that future clinical trials will combine sunitinib and conventional chemotherapeutic agents safely, by providing appropriate management of the AEs.
In conclusion, although the primary end point has not been met in this randomised phase II trial, the study has demonstrated that the addition of sunitinib can substantially improve response rate in metastatic gastric cancer in a second-line setting. Compared with docetaxel monotherapy, the addition of sunitinib was not associated with additional toxicity except stomatitis, diarrhoea, and HFS, and was not associated with more frequent treatment modification. The observation of some long-term responders indicates that the clinical development of sunitinib should still be pursued for treatment of gastric cancer. Especially with the advent of the personalised medicine, we should focus on exploring biomarkers that could identify those few gastric cancer patients who would dramatically respond to sunitinib.
Change history
23 January 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM 2003a SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther 2: 471–478
Abrams TJ, Murray LJ, Pesenti E, Holway VW, Colombo T, Lee LB, Cherrington JM, Pryer NK 2003b Preclinical evaluation of the tyrosine kinase inhibitor SU11248 as a single agent and in combination with ‘standard of care’ therapeutic agents for the treatment of breast cancer. Mol Cancer Ther 2: 1011–1021
Bang YJ, Kang WK, Kang YK, Kim HC, Jacques C, Zuber E, Daglish B, Boudraa Y, Kim WS, Heo DS, Kim NK 2002 Docetaxel 75 mg/m(2) is active and well tolerated in patients with metastatic or recurrent gastric cancer: a phase II trial. Jpn J Clin Oncol 32: 248–254
Bang YJ, Kang YK, Kang WK, Boku N, Chung HC, Chen JS, Doi T, Sun Y, Shen L, Qin S, Ng WT, Tursi JM, Lechuga MJ, Lu DR, Ruiz-Garcia A, Sobrero A 2010 Phase II study of sunitinib as second-line treatment for advanced gastric cancer. Invest New Drugs 29 (6): 1449–1458
Barone C, Basso M, Schinzari G, Pozzo C, Trigila N, D’Argento E, Quirino M, Astone A, Cassano A 2007 Docetaxel and oxaliplatin combination in second-line treatment of patients with advanced gastric cancer. Gastric Cancer 10: 104–111
Britten CD, Kabbinavar F, Hecht JR, Bello CL, Li J, Baum C, Slamon D 2008 A phase I and pharmacokinetic study of sunitinib administered daily for 2 weeks, followed by a 1-week off period. Cancer Chemother Pharmacol 61: 515–524
Christensen JG HC, Hollister BA 2008 Antitumor efficacy of sunitinib malate in concurrent and sequential combinations with standard chemotherapeutic agents in non-small cell lung cancer (NSCLC) nonclinical models. Proceedings of the 99th Annual Meeting of the American Association for Cancer Research. Philadelphia: PA, USA
de Jonge MJ, Dumez H, Kitzen JJ, Beuselinck B, Verweij J, Courtney R, Battista A, Brega N, Schoffski P 2011 Phase I safety and pharmacokinetic study of SU-014813 in combination with docetaxel in patients with advanced solid tumours. Eur J Cancer 47: 1328–1335
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG 2006 Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368: 1329–1338
Garcia-Donas J, Esteban E, Leandro-Garcia LJ, Castellano DE, del Alba AG, Climent MA, Arranz JA, Gallardo E, Puente J, Bellmunt J, Mellado B, Martinez E, Moreno F, Font A, Robledo M, Rodriguez-Antona C 2011 Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol 12: 1143–1150
Giuliani F, Gebbia V, De Vita F, Maiello E, Di Bisceglie M, Catalano G, Gebbia N, Colucci G 2003 Docetaxel as salvage therapy in advanced gastric cancer: a phase II study of the Gruppo Oncologico Italia Meridionale (G.O.I.M.). Anticancer Res 23: 4219–4222
Glimelius B, Ekstrom K, Hoffman K, Graf W, Sjoden PO, Haglund U, Svensson C, Enander LK, Linne T, Sellstrom H, Heuman R 1997 Randomized comparison between chemotherapy plus best supportive care with best supportive care in advanced gastric cancer. Ann Oncol 8: 163–168
Ilson DH 2007 Docetaxel, cisplatin, and fluorouracil in gastric cancer: does the punishment fit the crime? J Clin Oncol 25: 3188–3190
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D 2011 Global cancer statistics. CA Cancer J Clin 61: 69–90
Jung KW, Park S, Kong HJ, Won YJ, Lee JY, Park EC, Lee JS 2011 Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2008. Cancer Res Treat 43: 1–11
Kim JA, Lee J, Han B, Park SH, Park JO, Park YS, Lim HY, Kang WK 2011 Docetaxel/cisplatin followed by FOLFIRI versus the reverse sequence in metastatic gastric cancer. Cancer Chemother Pharmacol 68: 177–184
Lee JL, Ryu MH, Chang HM, Kim TW, Yook JH, Oh ST, Kim BS, Kim M, Chun YJ, Lee JS, Kang YK 2008 A phase II study of docetaxel as salvage chemotherapy in advanced gastric cancer after failure of fluoropyrimidine and platinum combination chemotherapy. Cancer Chemother Pharmacol 61: 631–637
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM 2003 In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9: 327–337
Moehler M, Mueller A, Hartmann JT, Ebert MP, Al-Batran SE, Reimer P, Weihrauch M, Lordick F, Trarbach T, Biesterfeld S, Kabisch M, Wachtlin D, Galle PR 2011 An open-label, multicentre biomarker-oriented AIO phase II trial of sunitinib for patients with chemo-refractory advanced gastric cancer. Eur J Cancer 47: 1511–1520
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, Solska E, Wilding G, Thompson JA, Kim ST, Chen I, Huang X, Figlin RA 2009 Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 27: 3584–3590
Nguyen S, Rebischung C, Van Ongeval J, Flesch M, Bennamoun M, Andre T, Ychou M, Gamelin E, Carola E, Louvet C 2006 Epirubicin-docetaxel in advanced gastric cancer: two phase II studies as second and first line treatment. Bull Cancer 93: E1–E6
Nishiyama M, Wada S 2009 Docetaxel: its role in current and future treatments for advanced gastric cancer. Gastric Cancer 12: 132–141
O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM 2003 SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101: 3597–3605
Park SH, Lim DH, Park K, Lee S, Oh SY, Kwon H, Kang JH, Hwang IG, Lee J, Park JO, Park YS, Lim HY, Kang WK 2011 A multicenter, randomized phase III trial comparing second-line chemotherapy (SLC) plus best supportive care (BSC) with BSC alone for pretreated advanced gastric cancer (AGC). J Clin Oncol 29 Suppl: Abstract 4004
Park SH, Kang WK, Lee HR, Park J, Lee KE, Lee SH, Park JO, Kim K, Kim WS, Chung CW, Im YH, Lee MH, Park CH, Park K 2004 Docetaxel plus cisplatin as second-line therapy in metastatic or recurrent advanced gastric cancer progressing on 5-fluorouracil-based regimen. Am J Clin Oncol 27: 477–480
Pyrhonen S, Kuitunen T, Nyandoto P, Kouri M 1995 Randomised comparison of fluorouracil, epidoxorubicin and methotrexate (FEMTX) plus supportive care with supportive care alone in patients with non-resectable gastric cancer. Br J Cancer 71: 587–591
Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, Chen JS, Horsch D, Hammel P, Wiedenmann B, Van Cutsem E, Patyna S, Lu DR, Blanckmeister C, Chao R, Ruszniewski P 2011 Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 364: 501–513
Robert F, Sandler A, Schiller JH, Liu G, Harper K, Verkh L, Huang X, Ilagan J, Tye L, Chao R, Traynor AM 2010 Sunitinib in combination with docetaxel in patients with advanced solid tumors: a phase I dose-escalation study. Cancer Chemother Pharmacol 66: 669–680
Tanaka M, Ma E, Tanaka H, Ioka A, Nakahara T, Takahashi H 2011 Trends of stomach cancer mortality in eastern Asia in 1950–2004: Comparative study of Japan, Hong Kong and Singapore using age, period and cohort analysis. Int J Cancer 130 4: 930–936
Van Cutsem E, Haller D, Ohtsu A 2002 The role of chemotherapy in the current treatment of gastric cancer. Gastric Cancer 5 Suppl 1: 17–22
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi E, Risse ML, Ajani JA 2006 Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24: 4991–4997
van Erp NP, Eechoute K, van der Veldt AA, Haanen JB, Reyners AK, Mathijssen RH, Boven E, van der Straaten T, Baak-Pablo RF, Wessels JA, Guchelaar HJ, Gelderblom H 2009 Pharmacogenetic pathway analysis for determination of sunitinib-induced toxicity. J Clin Oncol 27: 4406–4412
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE 2006 Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol 24: 2903–2909
Zhong H, Zhang Y, Ma S, Ying JE, Yang Y, Yong D, Hang X, Sun Q, Zhong B, Wang D 2008 Docetaxel plus oxaliplatin (DOCOX) as a second-line treatment after failure of fluoropyrimidine and platinum in Chinese patients with advanced gastric cancer. Anticancer Drugs 19: 1013–1018
Acknowledgements
This study was supported by SMC CRDP grant no. CRS109452. Pfizer provided sunitinib gratis, but the company was not involved in the accrual or analysis of the data, or in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yi, J., Lee, J., Lee, J. et al. Randomised phase II trial of docetaxel and sunitinib in patients with metastatic gastric cancer who were previously treated with fluoropyrimidine and platinum. Br J Cancer 106, 1469–1474 (2012). https://doi.org/10.1038/bjc.2012.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.100
Keywords
This article is cited by
-
Clinical Outcomes for Previously Treated Patients with Advanced Gastric or Gastroesophageal Junction Cancer: A Systematic Literature Review and Meta-Analysis
Journal of Gastrointestinal Cancer (2023)
-
Molecular targeted treatment and drug delivery system for gastric cancer
Journal of Cancer Research and Clinical Oncology (2021)
-
Targeted Therapies in Advanced Gastric Cancer
Current Treatment Options in Oncology (2020)
-
Targeted and novel therapy in advanced gastric cancer
Experimental Hematology & Oncology (2019)
-
Therapeutic impact of Nintedanib with paclitaxel and/or a PD-L1 antibody in preclinical models of orthotopic primary or metastatic triple negative breast cancer
Journal of Experimental & Clinical Cancer Research (2019)
