
Abstract
Background:
Exposure to ionising radiation is a well-established risk factor for multiple types of tumours, including malignant brain tumours. In the 1950s, radiotherapy was used to treat Tinea Capitis (TC) in thousands of children, mostly of North-African and Middle Eastern origin, during the mass migration to Israel. The over-representation of radiation-associated meningioma (RAM) and other cancers in specific families provide support for inherited genetic susceptibility to radiation-induced cancer.
Methods:
To test this hypothesis, we genotyped 15 families segregating RAM using high-density single-nucleotide polymorphism (SNP) arrays. Using the family-based association test (FBAT) programme, we tested each polymorphism and haplotype for an association with RAM.
Results:
The strongest haplotype associations were attained at 18q21.1 (P=7.5 × 10−5), 18q21.31 (P=2.8 × 10−5) and 10q21.3 (P=1.6 × 10−4). Although associations were not formally statistically significant after adjustment for multiple testing, the 18q21.1 and 10q21.3 associations provide support for a variation in PIAS2, KATNAL2, TCEB3C, TCEB3CL and CTNNA3 genes as risk factors for RAM.
Conclusion:
These findings suggest that any underlying genetic susceptibility to RAM is likely to be mediated through the co-inheritance of multiple risk alleles rather than a single major gene locus determining radiosensitivity.
Similar content being viewed by others

Main
A number of rare inherited cancer syndromes are typified by radiosensitivity, such as Gorlin and Li-Fraumeni syndromes (Gatti, 2001). Collectively, these diseases are, however, rare. Evidence that inherited sensitivity to radiation may have a more general genetic basis is provided by the observation that cancer patients and some of their first-degree relatives exhibit increased in vitro radiosensitivity compared with healthy controls (Roberts et al, 1999; Burrill et al, 2000).
Ionising radiation is the only environmental factor that has been shown unequivocally to be a causative factor for meningioma development (Sadetzki et al, 2005a; Bondy et al, 2008). During the mass migration to Israel in the 1950s, the Israeli authorities undertook a wide-scale campaign to eradicate Tinea Capitis (TC). The treatment included radiotherapy to the head area and was administered to children with TC in Israel and abroad, mainly in North-African and Middle Eastern countries, who were planning to immigrate to Israel. The therapeutic procedure followed the Adamson-Kienbock technique. The hair had been shaved and any remaining hair was removed through a waxing process. Subsequently, the scalp area was divided into five fields, each being treated on one of five consecutive days. The irradiation was done with a 75–100 kV superficial therapy X-ray machine. The children were exposed to 3.5–4.0 Gy for each field, at a focus skin distance of 25–30 cm. Most individuals received one course of radiation, but ∼9% of the patients received ⩾2 treatments (Werner et al, 1968).
A subgroup of children who were treated in Israel, including a group of 10 842 irradiated individuals with two matched nonexposed population and sibling groups (referred henceforth as the TC cohort), has been systematically followed for over 50 years for radiation sequelae. Radiation dosimetry was done for this cohort in the late 1960s using one of the original X-ray machines and a head phantom. These studies estimated the average dose to the brain at 1.5 Gy (s.d. 0.52, range 1.0–6.0 Gy). Doses were also calculated for different areas of the brain with the lowest average dose being for the back and front of the lower plane (mean 1.1 Gy, s.d. 0.37, range 0.71–4.30), whereas the highest dose was for the front of the upper plane (mean 1.8 Gy, s.d. 0.61, range 1.17–7.11) (Ron et al, 1988).
Although affecting <1% of the TC cohort, a marked increase in the risk of meningioma (ERR/Gy 4.63; 95% CI: 2.4–9.1) is one of the most prominent observations seen among the exposed individuals (Sadetzki et al, 2005a).
On the basis of the above-mentioned results, a law was established in Israel in 1994, for the purpose of compensating irradiated individuals who had developed specific diseases that were proven to be causally associated with the irradiation given as treatment for TC. The irradiated and nonirradiated cases and controls from the TC cohort, as well as irradiated cases who claimed compensation within the framework of the law, constitute the study population for nested case–control studies designed to assess interaction between ionising radiation and other environmental and genetic risk factors in the development of cancer (Sadetzki et al, 2005b; Flint-Richter and Sadetzki, 2007). These studies have demonstrated an over-representation of radiation-associated meningioma (RAM) and other radiation-associated cancers in specific families. This finding indicates that the occurrence of the tumour following the exposure is not a random event, and provides support for the hypothesis of inherited genetic susceptibility to radiation-induced cancers (Flint-Richter and Sadetzki, 2007).
The TC cohort is derived from a population that is characterised by high levels of linkage disequilibrium (LD). This allelic architecture affords enhanced power to localise and identify disease-causing alleles through association-based analyses especially if a restricted gene set underscores inherited susceptibility to RAM. In this study we report a search for RAM susceptibility alleles in TC families through an LD association-based analysis of genomewide single-nucleotide polymorphism (SNP) genotypes.
Subjects and Methods
Subjects
Our search for alleles predisposing to RAM was based on families ascertained through the TC studies. In total, 15 families in whom ⩾2 cases of RAM have been diagnosed among first-degree relatives were identified (Table 1). Of these families, 14 were identified from a larger epidemiological, genetic case–control study that included 160 RAM participants of whom 17 have reported on at least one sibling who was diagnosed with meningioma. However, out of these families, only 14 agreed to participate in the current study. More details on the methodology of this study have been previously published (Sadetzki et al, 2005b; Flint-Richter and Sadetzki, 2007). One additional family was recruited from the claim files, resulting in a total of 15 families. The target study population included 120 individuals (40 RAM, 14 healthy irradiated, 49 healthy nonirradiated, 9 irradiated with other cancer and 8 nonirradiated with other cancer); the number of siblings in each family ranged from 5 to 12. The age at diagnosis for the RAM patients ranged from 35 to 69 years (mean 48.7±9.2). Validation for irradiation status and for tumour pathology was performed for all of these family members, using medical records for pathology verification and a set of criteria that were used in previous studies (Sadetzki et al, 2005b) for irradiation verification.
Biological specimens were collected from 71 individuals; however, the final genetic analysis was based on 65 samples because only DNA extracted from peripheral blood (n=66, 27 RAM) was used and 1 DNA was excluded because of having an overall call rate of <95% (Table 1).
Ethics
Collection of blood samples and clinicopathological information from subjects was undertaken with informed consent and relevant ethical review board approval in accordance with the tenets of the Declaration of Helsinki.
Genotyping
DNA was extracted from EDTA-venous blood samples using conventional methods and quantified using PicoGreen (Invitrogen, Carlsbad, CA, USA). Genotyping was conducted using Illumina 610Quad arrays according to the manufacturer's protocols (Illumina San Diego, CA, USA). To ensure quality of genotyping, a series of duplicate samples was genotyped, resulting in 99.99% concordant calls. We excluded SNPs from analysis if they failed one or more of the following thresholds: GenCall scores <0.25; overall call rates <95%; minor allele frequency (MAF) ⩽0.01; outlying in terms of signal intensity or X : Y ratio; discordance between duplicate samples; and, for SNPs with evidence of association, and poor clustering on inspection of X : Y plots.
Statistical analyses
The primary analysis was for association of individual SNPs with the binary trait of RAM, using the family-based association test (FBAT) programme (Horvath et al, 2001), and for haplotypes, using the haplotype extension (HBAT) of the FBAT programme (Horvath et al, 2004). FBAT is a generalised version of the classical transmission–disequilibrium test, which can be applied to any type of nuclear family (Laird et al, 2000), thus avoiding the issue of population admixture that is a commonly encountered in case–control study designs. In the FBAT programme, the additive model was used.
Haplotype analyses were performed using sliding window sizes of 12 contiguous markers. Haplotype frequencies for each individual were estimated using an expectation-maximisation (EM) algorithm. The minimum haplotype frequency was set at 0.01, and haplotypes with frequencies below this threshold were combined into a single group.
Because of the large number of multiple tests performed, we used the Benjamini and Hochberg correction (Benjamini et al, 2001) for multiple testing, which is a method for controlling the false discovery rate, to adjust the haplotypic P-values. This correction consists of ranking all the P-values, from smallest to largest, and adjusting each by multiplying by the total number of tests and dividing by the rank of that P-value. All test statistics with rank less than the test statistic with the largest rank for which the corrected value is less than the desired error rate (e.g., 0.05) are significant.
Mutational analysis
A search for mutations in the coding regions and splice sites of all isoforms of CTNNA3 and LRRTM3, as annotated by GRChB37, was performed by sequencing amplified PCR fragments using BigDye Terminator chemistry implemented on an ABI 3730xl sequencer (Applied Biosystems, Carlsbad, CA, USA). PCR primers were designed using Primer 3 software and are given in Supplementary Table 1. Sequence traces were aligned and compared with the gene consensus sequence using Mutation Surveyor (Version 3.2; SoftGenetics, State College, PA, USA). Two in silico algorithms, PolyPhen (http://genetics.bwh.harvard.edu/pph/) and SIFT (http://sift.jcvi.org), were used to predict the putative impact of missense variants on protein function. Scores were classified as tolerated, borderline or deleterious according to the proposed criteria.
Results
Illumina 610Quad SNP genotypes were obtained for all 66 samples genotyped. Before conducting association-based analyses, we subjected the SNP data set to rigorous quality control in terms of excluding samples and SNPs with poor call rates. As mentioned previously, one sample was excluded because of having a call rate of <95%; the remaining samples had average call rates across all SNPs of >99%. Thus, the final analysis was based on 65 samples. Following this, we critically evaluated the data set for ancestral differences by principal component analysis (Figure 1). Although minor differences were apparent, all individuals genotyped were relatively ancestrally comparable. Thus, without introducing significant systematic bias we considered the data set to be uniform to maximise power to detect important associations under the assumption of homogeneity and an ancestral risk haplotype for RAM. In all analyses we treated individuals with RAM as affected and all other family members as of unknown phenotype.

Principal component analysis of SNP genotypes showing the extent of ethnic variability in the TC cohort. The first two principal components of the analysis were plotted. HapMap CEU (Caucasian) individuals are denoted by grey triangles, CHB (Chinese Han Beijing)+JPT (Japanese in Tokyo) by grey diamonds, YRI (Yoruba) by grey squares and TC cohort individuals are plotted in black.
The median distance between the 575 272 autosomal SNPs in the Illumina 610Quad arrays was ∼2.7 Kb and ∼88% of the genome was within 10 Kb of a SNP marker. In this study, the heterozygosity of markers was ∼94%, hence almost as many SNPs present on that array are heterozygous in this Jewish population as in the general Caucasian population.
We systematically interrogated haplotypes defined by a varying number of SNPs. Haplotypes defined by >12 SNPs proved too computationally intensive to recover on a genomewide basis. We therefore restricted our search for disease-associated risk locus on the basis of 12 SNP haplotypes.
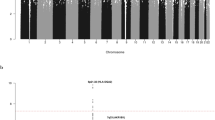
This analysis provided results on the association between RAM and 41 414 haplotype tests across the genome (Figure 2). In all, 66 haplotype tests provided evidence for an association between genotype and RAM at P<0.001 (Table 2) including multiple haplotypes on chromosomes 18 and 10. The strongest associations were shown at 18q21.1 (P=7.5 × 10−5), 18q21.32 (P=2.8 × 10−5) and 10q21.3 (P=1.6 × 10−4).

Manhattan plot of genomewide haplotype test P-values for the association between haplotypes and RAM. The –log10 P-values (y axis) are presented at their chromosomal positions (x axis).
A number of genes map to the 18q21.1 region of association including PIAS2, KATNAL2, TCEB3CL, TCEB3C, TCEB3B and DKfZp667C165, whereas the 18q21.31 region is bereft of genes. In contrast to the other associations, the 10q21.3 signal was characterised by a large number of neighbouring haplotype associations; eight providing evidence for an association at P<0.001. These haplotypes all mapped within a 2 Mb region of 10q21.3 and all annotate the catenin (cadherin-associated protein), α-3 (CTNNA3) gene. Among the top 66 associations, we identified only two other genes that were annotated by multiple haplotype tests showing evidence for an association at P<0.001. Specifically, TMC1 on 9p21.13 and PCDH15 on 10q21.1 were captured two and three times, respectively, by haplotype associations (Table 2).
The CTNNA3 is part of the Wnt signalling pathway and, although speculative, CTNNA3 represents an attractive basis for susceptibility given the role of dysfunctional Wnt signalling in radiosensitivity. In view of this, we explored the possibility that a common or restricted set of coding sequence changes in CTNNA3 might underscore the 10q21.3 association. For completeness we also screened the leucine rich repeat transmembrane neuronal 3 (LRRTM3) gene that maps internally within CTNNA3 (Figure 3).

Haplotype –log10 P-values for the 10q21.3 region. Beneath the plot are the five isoforms of CTNNA3 and two of LRRTM3 with exons shown as black blocks and UTRs as empty blocks, annotated as per Supplementary Table 2.
Nine sequence changes within coding sequence were identified in the same 65 individuals whose DNA passed QC in the genomewide stage. These included five polymorphic variants documented in dbSNP (four in CTNNA3 and one in LRRTM3) and four novel changes (three and one in CTNNA3 and LRRTM3, respectively). Seven of the variants identified were missense changes, six in CTNNA3 and one in LRRTM3. None of the missense changes identified were confined to individuals with a RAM phenotype (Supplementary Table 2). Furthermore, none of the sequence changes were predicted to impact on the functionality of the expressed protein.
Discussion
The TC cohort is unique, and has allowed us to recently assess the impact of environmental and inherited risk factors on tumour development in those exposed to ionising radiation (Bondy et al, 2008). In contrast to the rarity of familial meningioma in the general population, 17 families within the TC study had two or more members affected with RAM, equating to sibling relative risk of ∼20-fold. As the doses of therapeutic radiation administered to individuals within the TC cohort were similar (interquartile range (25–75%) 127–153 cGy), it has raised the possibility that the impact of ionising radiation on cancer risk is in part a consequence of genetic susceptibility conferred by low penetrance genes.
To provide evidence for this hypothesis and identify a RAM-associated disease locus, we have conducted an associated analysis using high-density SNP genotyping. The SNP LD mapping strategy employed in this study has relied on the comparatively large regions of LD that encompass founder mutations segregating in the Jewish population. This simple study design strategy and using DNAs from a small number of people has previously been successfully used to localise a susceptibility gene for Bloom's syndrome (Mitra et al, 2004).
Predicated on the assumption of inherited predisposition, our study provides insight into the possible architecture of genetic susceptibility to RAM. Over a range of gene frequencies of 0.001 to 0.05, and stipulating a false positive rate of 0.0001, our study had high power (>70%) to identify a disease-associated haplotype contributing >30% of the excess risk assuming a simple genetic model of familial aggregation. Although assuming an effect size of 30% is high for many complex traits as previously articulated, an assumption on which our study was predicated is that RAM is primarily a consequence of major gene susceptibility and because of the restricted ethnicity allelic heterogeneity is limited.
An alternative model of RAM is that this phenotype is a consequence of a complex-polygenic basis. Failure to unambiguously identify a single locus is thus entirely compatible with the latter model of disease susceptibility, whereby disease risk is mediated by alleles conferring more modest effects, possibly through the consequence of the co-inheritance of multiple low-risk variants. Under this model, we would have had only very limited power to identify a disease-causing locus, stipulating a P-value of 1 × 10−6 to ensure genomewide significance. At the lower significance threshold, our analysis does provide some evidence to support the involvement of a number of genes in the aetiology of RAM; specifically, the gene encoding CTNNA3 that is captured by the 10q21.3 haplotypes. This gene is of specific interest as it is part of the Wnt signalling pathway, which has been related to cancer development and neurodegeneration. Several components of the Wnt pathway have been implicated in carcinogenesis and are best known to be involved in colorectal, lung, prostate, breast and skin cancers (Behrens et al, 2009; Lai et al, 2009). Moreover the CTNNA3 gene contains a fragile site of potential interest in terms of genomic instability as there is evidence suggesting that it may function as a tumour suppressor (Smith et al, 2006).
Although there is evidence for inherited susceptibility to radiosensitivity outside the context of a restricted set of syndromes, it is primarily derived from in vitro data. However, the phenotype radiosensitivity is relatively prosaic, and establishing a relationship between genotype and sensitivity is inherently problematic as multiple clinical end points can be considered, many of which are ill defined. To obviate this, we have made use of a unique cohort and have sought to establish a relationship between constitutional genotype and cancer risk.
Failure in our study to unambiguously identify a single high-risk locus provides evidence for a model of inherited susceptibility to radiosensitivity based on the co-inheritance of multiple low-risk variants. Although individually such loci only confer small effects, it is likely that they act multiplicatively, exerting relatively profound effects in a small proportion of the population.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Behrens MI, Lendon C, Roe CM (2009) A common biological mechanism in cancer and Alzheimer's disease? Curr Alzheimer Res 6 (3): 196–204
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125 (1-2): 279–284
Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il′yasova D, Kruchko C, McCarthy BJ, Rajaraman P, Schwartzbaum JA, Sadetzki S, Schlehofer B, Tihan T, Wiemels JL, Wrensch M, Buffler PA (2008) Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer 113 (7 Suppl): 1953–1968
Burrill W, Barber JB, Roberts SA, Bulman B, Scott D (2000) Heritability of chromosomal radiosensitivity in breast cancer patients: a pilot study with the lymphocyte micronucleus assay. Int J Radiat Biol 76 (12): 1617–1619
Flint-Richter P, Sadetzki S (2007) Genetic predisposition for the development of radiation-associated meningioma: an epidemiological study. Lancet Oncol 8 (5): 403–410
Gatti RA (2001) The inherited basis of human radiosensitivity. Acta Oncol 40 (6): 702–711
Horvath S, Xu X, Laird NM (2001) The family based association test method: strategies for studying general genotype--phenotype associations. Eur J Hum Genet 9 (4): 301–306
Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM (2004) Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol 26 (1): 61–69
Lai SL, Chien AJ, Moon RT (2009) Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res 19 (5): 532–545
Laird NM, Horvath S, Xu X (2000) Implementing a unified approach to family-based tests of association. Genet Epidemiol 19 (Suppl 1): S36–S42
Mitra N, Ye TZ, Smith A, Chuai S, Kirchhoff T, Peterlongo P, Nafa K, Phillips MS, Offit K, Ellis NA (2004) Localization of cancer susceptibility genes by genome-wide single-nucleotide polymorphism linkage-disequilibrium mapping. Cancer Res 64 (21): 8116–8125
Roberts SA, Spreadborough AR, Bulman B, Barber JB, Evans DG, Scott D (1999) Heritability of cellular radiosensitivity: a marker of low-penetrance predisposition genes in breast cancer? Am J Hum Genet 65 (3): 784–794
Ron E, Modan B, Boice Jr JD, Alfandary E, Stovall M, Chetrit A, Katz L (1988) Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med 319 (16): 1033–1039
Sadetzki S, Chetrit A, Freedman L, Stovall M, Modan B, Novikov I (2005a) Long-term follow-up for brain tumor development after childhood exposure to ionizing radiation for tinea capitis. Radiat Res 163 (4): 424–432
Sadetzki S, Flint-Richter P, Starinsky S, Novikov I, Lerman Y, Goldman B, Friedman E (2005b) Genotyping of patients with sporadic and radiation-associated meningiomas. Cancer Epidemiol Biomarkers Prev 14 (4): 969–976
Smith DI, Zhu Y, McAvoy S, Kuhn R (2006) Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 232 (1): 48–57
Werner A, Modan B, Davidoff D (1968) Doses to brain, skull and thyroid, following x-ray therapy for Tinea capitis. Phys Med Biol 13 (2): 247–258
Acknowledgements
We thank all individuals who participated in this study. Work at the Institute of Cancer Research was supported by grants from Cancer Research UK (C1298/A8780 and C1298/A8362).
Web addressesThe URLs for data/software presented herein are as follows:
FBAT: http://www.biostat.harvard.edu/~fbat/
Primer 3: http://primer3.sourceforge.net/
NCBI dbSNP: http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/Omim/
GoldenHelix: http://www.goldenhelix.com/SNP_Variation/HelixTree/
Ilumina: http://www.illumina.com/
Haploview: http://www.broadinstitute.org/haploview/haploview
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hosking, F., Feldman, D., Bruchim, R. et al. Search for inherited susceptibility to radiation-associated meningioma by genomewide SNP linkage disequilibrium mapping. Br J Cancer 104, 1049–1054 (2011). https://doi.org/10.1038/bjc.2011.61
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.61
