
Abstract
Cell states in hematopoiesis are controlled by master regulators and by complex circuits of a growing family of RNA species impacting cell phenotype maintenance and plasticity. Circular RNAs (circRNAs) are rapidly gaining the status of particularly stable transcriptome members with distinctive qualities. RNA-seq identified thousands of circRNAs with developmental stage- and tissue-specific expression corroborating earlier suggestions that circular isoforms are a natural feature of the cell expression program. CircRNAs are abundantly expressed also in the hematopoietic compartment. There are a number of studies on circRNAs in blood cells, a specific overview is however lacking. In this review we first present current insight in circRNA biogenesis discussing the relevance for hematopoiesis of the highly interleaved processes of splicing and circRNA biogenesis. Regarding molecular functions circRNAs modulate host gene expression, but also compete for binding of microRNAs, RNA-binding proteins or translation initiation and participate in regulatory circuits. We examine circRNA expression in the hematopoietic compartment and in hematologic malignancies and review the recent breakthrough study that identified pathogenic circRNAs derived from leukemia fusion genes. CircRNA high and regulated expression in blood cell types indicate that further studies are warranted to inform the position of these regulators in normal and malignant hematopoiesis.
Similar content being viewed by others

Introduction
Circular RNAs (circRNAs) are covalently closed RNA molecules, in which the 3′- and 5′-ends are linked in a non-collinear way by a process called back-splicing.1 Unlike in linear RNA splicing, a splice donor site is joined to a splice acceptor site upstream in the primary transcript, yielding a circRNA.1 CircRNAs can be formed by circularization of a single exon, two or more exons,2 both exons and intron sequences (exon–intron circRNA, EIcirRNA)3 or intronic sequences only (circularized intron RNA; ciRNA)4 (Figure 1). Several circular isoforms can be produced from a given gene, and different circRNAs from the same gene may show distinct expression profiles, as reported for circSTAU2a and circSTAU2b.5

Linear and circRNAs. CircRNAs are produced by backsplicing, and combinations of exons and introns give rise to different products, including single circularized exons, circRNAs formed by two or more exons, by exon and retained intron sequences (EI-ciRNAs) and by intronic sequences only.
Circularity confers specific properties to circRNA: they are highly stable, resistant to RNAse R and have longer half-lives compared with linear RNAs6, 7 and tend to accumulate in cells with a low proliferation rate.8 Detection of circRNA in human body fluids such as plasma9 and saliva10 indicates circRNAs as potential disease biomarkers.
The first description of circRNA dates back to several decades ago. Recently, circRNAs were relaunched by RNA-seq-based studies as an RNA species with high relevance for molecular biology and molecular oncology, and today over 10 000 human circRNAs have been identified.11, 12, 13, 14, 15, 16, 17
CircRNAs are non-poly-adenylated and coincidental discovery of circRNA in the past can be attributed to RNA extraction methods that mainly used polyA selection. Naturally occurring single-stranded covalently closed RNA molecules were first described in plant viroids18 and were valued for their peculiar structure that allows for rolling circle replication.1 A few studies in the nineties reported non-canonical splicing with scrambled exons of candidate tumor suppressor gene DCC,19 ‘missplicing’ of ETS1 transcripts20 and murine Fmn,21 and exon circularization in nuclear extracts.22 Moreover, whereas early in development the mRNA of therian SRY is translated into the protein that triggers the sex-determining transcriptional cascade, in adults SRY transcripts are found as cytoplasmic circular SRY (cSRY) not particularly bound to polysomes23 and later proven to efficiently sponge miR-138.24 Other primary RNAs were found to be processed into circRNA isoforms such as MLL (KMT2A),25 ETS1,26 CYP2C18,27 SLC8A1(ref. 28) and dystrophin (DMD)29 transcripts. Examples of circRNAs corresponding to linear noncoding RNA, as well as antisense RNA were also detected.30, 31
Most of the above-mentioned studies postulated relevant biological functions for circular RNAs but were only confined to certain genes. In any case, shortly after publication of circular forms of an INK4/ARF-associated non-coding RNA30 numerous studies embarked on transcriptome wide circRNA analysis showing developmental stage- and tissue-specific expression, and specific regulatory roles for circRNAs were suggested.2, 13 These new data triggered the interests of the scientific community resulting in the development of molecular methods to study circRNAs and of microarray platforms to measure expression levels of circRNAs (Figure 2), as well as the implementation of bioinformatics software to detect and discover circRNAs from RNA-seq data (Table 1) posing the basis for further experimental circRNA characterization, and for circRNA quantification and differential expression testing. Moreover, several available circRNA databases and web resources (Table 2) could be rather useful to explore putative circRNA interactions and functions.

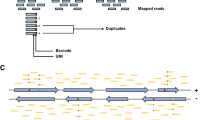
Molecular methods for circRNA detection, validation and study. (a) CircRNA detection from RNA-seq data grounds on the identification of sequence reads encompassing the backsplice junction; (b) Backsplice reads map to the genome in chiastic order (two segments of a single read align separately in reverse order) due to the backsplicing in circRNAs biogenesis. (c) Convergent primers (white arrows) designed in adjacent spliced exons amplify both linear and circular isoforms, whereas primers that are divergent in the linear transcripts (black arrows) can be used to specifically amplify the circular isoform; (d) PolyA enrichment protocols deplete circRNAs, whereas ribosome depletion and RNAse R protocols enrich circRNAs; (e) RNAse R digestion before reverse transcription–PCR lowers the amount of false-positive amplicons facilitating circRNA validation; (f) Gel Trap electrophoresis allows isolate the circular and linear fractions of the input RNA, as circRNAs are hold in the well; (g) Two-dimensional acrylamide gel electrophoresis separates the circular RNA fraction in an off-diagonal curve; (h) RNA migration in agarose gel before and after a mild RNAse H treatment resulting in a single cut per molecule shows that circular molecules bearing a ‘backsplice’ junction are discriminated from linear ones deriving from a duplication event, as only circRNA results in a single band after being cut once (f–h re-elaborated from56).
Several studies on circRNAs in blood cell types and hematologic malignancies were recently conducted and will be discussed below.
CircRNA biogenesis
Back-splicing
As anticipated, circRNA loops are generated by back-splicing from immature RNA, where ends are joined in a non-collinear way. CircRNAs are derived from Pol II transcripts just like linear transcripts. Back-splicing requires the spliceosomal machinery32, 33, 34 as revealed by treatment of HeLa cells with a splice inhibitor followed by nascent RNA purification.
In the majority of cases, the generation of circRNA happens at the expense of their corresponding mRNA isoforms, and is characterized by the usage of canonical splice sites that precisely flank head-to-tail junctions of circular transcripts. In some cases, transcripts of specific genes are predominantly spliced into the circular isoform.11, 12 Ashwal-Fluss et al.32 demonstrated that circularization and splicing of linear forms compete against each other. Kelly et al.35 confirmed a direct correlation between exon skipping and circularization. Thus, circRNA biogenesis and regulation of mRNA production are tightly linked.
Cis regulatory features
CircRNA-forming exons are often flanked by particularly long introns, possibly reducing splicing efficiency.12, 16 Moreover, in humans these long introns are markedly enriched in ALU repeats,12 and complementary sequences in introns are involved in specific folding of primary transcripts that favor circularization.32 In the Sry gene, the activation of an upstream promoter triggers the synthesis of a primary transcript containing inverted repeats needed for circRNA production.36 As drosophila RNA circularization does not appear to be driven by structural complementarity of exon-bordering sequences but only determined by the length of exon-flanking introns, inverted repeats alone do not fully explain the production of circRNAs in eukaryotes. In addition, regulation of the dynamic expression of circRNAs in different cell types is likely also dependent on control by trans-acting factors.37
Trans-acting factors
Besides the role of flanking sequence elements, introns encasing circRNAs are highly enriched in RNA A-to-I editing events.12 In fact, knockdown of RNA-editing enzyme ADAR1 upregulated circRNA expression, favoring a mechanism of circRNA biogenesis whereby ADAR1 antagonizes circRNA expression by melting stems of RNA–RNA interactions within introns that putatively promote circularization.12
The Muscleblind (MBL) family of splicing factors was also shown to take part in the regulation of circRNA production by binding specific intronic sites flanking circularized exons.32 Intriguingly, in the fly circRNA isoform expression of MBL itself is regulated by MBL protein. Decrease of circRNA expression after MBL knockdown supports a circRNA-promoting role for MBL proteins.32
In addition, RNA-binding protein (RBP) Quaking (QKI) regulates the formation of circular RNAs.38 QKI dimers bind to specific bipartite sequences termed QKI response elements that are present in many RNAs, including coding mRNAs and primary miRNAs. Conn et al.38 investigated the role of QKI in promoting circRNA biogenesis in transforming growth factor-beta-induced epithelial–mesenchymal transition of the epithelial HMLE cell line, demonstrating that the knockdown of the QKI-5 isoform specifically decreases the formation of circRNAs, and that insertion of synthetic QKI response elements in introns mediates circRNA formation. Metabolic tagging of nascent RNAs with 4-thiouridine has been used to study the link between nascent circRNA processing and transcription17 showing that the efficacy of circRNA processing from primary transcripts is extremely low. This study also clarified that circRNAs are largely processed post-transcriptionally and confirmed that circRNAs are stable, being thus abundant at a steady-state level and tending to accumulate particularly in cells with low proliferation rates.8
Backsplice as a new form of alternative splicing
Alternative RNA splicing is a complex tightly regulated phenomenon. Since the discovery of split genes robust knowledge was built on splicing prevalence, on complexity of splicing patterns and on molecular mechanisms that determine, regulate or change splicing, including RNA–protein interactions (splicing factors with in cis regulatory sites termed silencers or enhancers), RNA–RNA base-pairing interactions involving both in trans acting RNAs and in cis secondary structure formation, and also chromatin-based effects.39
Disease-causing mutations occur in splice sites or in regulatory elements, as well as in genes that encode splicing factors (U2AF1, SRSF2, SF3B1 and ZRSR2), and there is much interest in developing antisense oligonucleotides to control splicing patterns and using genome editing to correct disease-causing splicing defects. Alternative splicing is highly and commonly deregulated in cancer cells40, 41, 42 and specifically impacts prognosis and disease course of myeloid malignancies, including chronic lymphocytic leukemia, acute lymphoblastic leukemia (ALL), acute myeloid leukemia, and myeloproliferative neoplasms.43, 44, 45, 46
As circRNA biogenesis and splicing are interleaved processes, it can be hypothesized that mutations of splicing factors and/or alterations of regulatory elements have an impact on circRNA biogenesis. RBP involved in circRNA biogenesis might drive developmental regulation of circRNA formation and show deregulation in disease. Distinct expression levels of ADAR1, MBNL1 and QKI in normal bone marrow compared with B-cell leukemia subtypes (Figure 3) encourage investigations as also subtle expression variations of ADAR1 were shown to be relevant for RNA circularization.5

Expression variation of enzymes involved in circRNA expression. Gene expression intensities of ADAR1, MBNL1 and QKI in samples of normal bone marrow and six B-cell leukemia subtypes carrying specific genetic aberrations (according to Haferlach et al.87); expression data obtained with HG-U133 Plus 2.0 (Affymetrix, Santa Clara, CA, USA).
Alternative splicing is a key mechanism through which fundamental processes during hematopoiesis are regulated,47 posing the basis to interpret the consequences of genetic variation. Similarly, there is high demand to study circRNA in normal hematopoiesis, to connect biogenic mechanisms with biological functions of circRNAs, to accumulate fundamental knowledge needed to understand disease mechanisms and to inform strategies for therapeutic intervention.
CircRNA degradation
As circRNAs are endogenous cell products one might ask which endogenous mechanisms cells have to dispose of circRNA. In general, RNA is degraded by the exosome, a multiprotein complex that reminiscently of the proteasome forms a chamber with helicase activity, which unfolds and then degrades RNA. The degradation is prevalently exoribonucleolytic from the 3′-end, but the exosome catalytic subunit RRP44 also has endonuclease activity.48
According to available evidences circRNAs are not degraded by treatments (as tobacco acid pyrophosphatase plus terminator 5′‐phosphate dependent exonuclease or highly processive 3′- to 5′- exoribonuclease RNAse R digestion) that normally degrade linear RNA with free ends.31 Regarding degradation, in general miRNAs can regulate cleavage of circRNAs. The better-characterized path toward degradation of a circRNA is that of CDR1-as (circular antisense transcript deriving from cerebellar degeneration-related protein 1 locus) that even presenting multiple miR-7 binding sites is completely resistant to miR-7-mediated degradation and also resistant to miR-769-mediated degradation, whereas the binding of miR‐671 to CDR1-as directs Ago2‐slicer‐dependent cleavage.31
Undoubtedly, our understanding of the regulation of circRNA turnover and endogenous degradation mechanisms is limited. We can hypothesize that not only the deregulation of circRNA synthesis but also its degradation are biologically relevant. For instance, a lack of cleavage might result in undesired circRNA accumulation.
CircRNA conservation and molecular functions
CircRNAs are evolutionarily conserved
CircRNAs were described in many eukaryotes from yeast to humans49 and resulted very conserved at the nucleotide level: Memczak et al.13 analyzed sequence conservation within circRNAs and showed that 223 human circRNAs with conserved circularization in mice were significantly more conserved in the third codon positions than exons not engaged in circular forms. CircRNAs are also depleted of polymorphisms in miRNA-binding sites.50 Beyond apparent sequence conservation, both paralogous and orthologous gene pairs have been reported to express circular transcripts: human HIPK2 and HIPK3, as well as murine Hipk2/3 produce circRNAs.12 Also conservation of circRNAs in terms of exonic sequences, bordering intronic sequences, precise backsplice junctions and expression patterns in mammals and to some extent in Drosophila has been recently reported.5 The above indications of evolutionary preservation point to a central position of circRNAs in core biological processes.
CircRNAs are seldom translated
It has previously been shown that eukaryotic ribosomes can initiate translation on circRNAs containing internal ribosome entry site elements (Figure 4) producing long-repeating polypeptides in the presence of a continuous open reading frame.51, 52 Efficient circRNA translation can occur in HEK293(ref. 34) and Hela cells.53 Intriguingly, a circRNA can be a sort of ‘Mobius strip’ with translation generating proteins either recurrently, or variably depending on whether or not the sequence length is a multiple of three nucleotides. A small viroid circRNA directly translated through three completely overlapping open reading frames shifting to a new reading frame at the end of each round has been reported as a natural supercompact ‘nanogenome’.54

CircRNA functions. Elucidated circRNA functions include the ability to sponge miRNAs thus regulating the silencing of canonical targets (for example, ciRS-7/CDR1-as harbors 63 binding sites for miR-7) and participating to ceRNA networks; similarly circRNAs could decoy RBP ultimately regulating the functions in which RBP are implicated (for example, circ-Foxo3 forms a ternary complex with p21 and CDK2 arresting cell cycle progression); circRNAs can also regulate in cis the expression if the gene from which they derive through interactions with the U1 RNA in the U1 RNP in the nucleus (for example, circEIF3J); moreover, circRNAs harboring an IRES could be translated to produce peptides or compete with mRNA translation (for example, circFMN contains an active translation start site not leading to the protein synthesis).
Even if in principle circRNAs can be translated, the majority of recently discovered and characterized circRNAs seem to have limited coding potential: seldomly associated neither with messenger ribonucleoprotein particles nor with translationally active polyribosomes, suggesting that circRNAs, as a species, are unlikely to be translated into peptides.55 The fact that in the same study mass spectrometry failed to identify peptides encoded by backsplice junctions of circRNAs could be due to low sensitivity or to the position of open reading frames outside junctions and does not rule out that part of circRNAs can be translated in some cell types and/or conditions.
A circRNA produced by murine Fmn21 contains an active translation start site not leading to protein synthesis. In this way, the circular form competes with the linear mRNA both impacting on the linear transcript abundance and providing an ‘mRNA trap’ that can sequester proteins of the translation initiation complex. Also, Jeck and Sharpless56 uncovered that many single-exon circRNAs contain a translation start site,57 further exemplifying this mechanism of protein expression regulation by circRNAs.
CircRNAs are efficient miRNA sponges and participate to competing endogenous RNA networks
As demonstrated by several studies, circRNAs with multiple miRNA-binding sites are efficient miRNA sponges that participate in the regulation of specific cellular pathways (Figure 4).24, 58
CDR1-as harbors 63 conserved binding sites for miR-7 displaying high miRNA-binding capacity and miRNA antagonist activity in the brain.13, 24 Following this functional description CDR1-as was renamed circular transcript ciRS-7 (circular RNA sponge for miR-7) or called CDR1-as/ciRS-7. Notably, the circRNA is completely resistant to miR-7-mediated target destabilization and strongly suppresses miR-7 activity, resulting in increased levels of miR-7 targets, including EGFR and IRS2.
The previously mentioned cSRY decoys miR-138, for which it displays 16 target sites.12, 24 Circ-ITCH, a circRNA downregulated in carcinomas, was demonstrated to be a sponge of miR-7, miR-17 and miR-214, by increasing the level of ITCH and ultimately inhibiting the Wnt/β-catenin pathway.58, 59 Another circRNA (hsa_circ_001569) that acts as a sponge for miR-145 upregulates its targets enhancing cell proliferation and invasion of colorectal cancer.60
Recently, specific circRNA–miRNA axes have been shown to regulate cancer-related processes. CircRNAs can have both cancer-promoting and -suppressing roles, depending on the molecular circuits in which they are involved and on the role of the interactors.58, 59, 61 CircRNAs can exhibit anticancer effects: as synthetic circular sponges displayed superior anticancer activities compared with the linear sponges, RNA circles open new ways to deliver miRNA sponges with persistent effects.26
CircRNAs like linear isoforms can act as competing endogenous RNAs (ceRNAs) that decoy miRNAs and indirectly regulate protein-coding gene expression (Figure 4).62 ceRNAs are implied in the progression of cancer and impact on cancer hallmarks.57 Being resistant to miRNA-mediated degradation circRNA can presumably also tether RISC components depriving the cellular pool of both miRNAs and RISC effectors.58
Following the observations of circRNAs acting as ceRNAs it has been asserted that such a mechanism may be common to all circRNAs. The latter finds support from previously mentioned shortage of polymorphisms in circRNAs’ putative miRNA-binding sites. Other studies showed instead that only a minority of expressed circRNAs present multiple binding sites for specific miRNAs, and according to their observations circRNAs are, in general, not bound to miRNA-loaded Argonaute proteins.56, 63 In addition, argonaute co-immunoprecipitation experiments did not indicate an appreciable enrichment of circRNA-derived exons among argonaute family-bound transcripts, which would be expected if circRNAs were prevalently acting as ceRNAs. See Thomson et al.64 for a review on evidences and open questions on endogenous miRNA sponges. According to available data, supported by recent findings3, 59 we conclude that some circRNAs can act as ceRNAs, whereas others may be involved in a variety of other molecular mechanisms.
Interactions with RBPs
The decoy activity of circRNAs could be important also for RBPs (Figure 4). CircRNAs like linear RNAs may interact with RBPs in a sequence-specific and structural motif determined way. CircRNAs could function to store, sort or localize RBPs. Recently, the interaction between Foxo3 circular RNA and specific proteins was shown to delay cell cycle progression.65 Foxo3 is a forkhead box O transcription factor and may behave as a tumour suppressor protein that limits cell proliferation and induces apoptosis and is frequently altered in cancer, shown to be deleted in lymphomas (diffuse large B-cell lymphoma), and translocated with MLL in leukemia.66 In healthy cells high circ-Foxo3 expression was found to be associated with cell cycle progression. Silencing endogenous circ-Foxo3 promoted cell proliferation, whereas ectopic expression of circ-Foxo3 repressed cell cycle progression by binding to cell cycle proteins cyclin-dependent kinase 2 (CDK2) and cyclin-dependent kinase inhibitor 1 (or p21). Normally, CDK2 interacts with cyclin A and cyclin E to facilitate cell cycle entry, while p21 inhibits these interactions and arrests cell cycle progression. The formation of the circ-Foxo3–p21–CDK2 ternary complex arrests the function of CDK2 and blocks cell cycle progression. This study identified an oncogenic function of a circRNA and indirectly demonstrated that circRNAs can have distinct functions with respect to that of protein products encoded by the same gene.65
Cis regulation of gene expression by circRNAs
Another line of evidence reported cis-regulatory roles for specific circRNAs. Exon–intron circRNAs derived from circularization of RNA with intron retention were identified as a subclass of ciRNAs, enriched in the nucleus, associated with Pol II.3 Further analyses of two exon–intron circRNAs (circEIF3J and circPAIP2) showed interactions with Pol II, U1 snRNP and parental gene promoters through sequence complementarity between the U1 snRNA and an U1-binding site, which eventually promote the transcription of the gene from which they derived (host genes), triggering a positive-feedback loop.
Zhang et al.4 described circular intronic RNAs (ciRNAs) that were found to accumulate in human cells due to a failure in debranching and showed that knockdown of ciRNAs reduced expression of their host genes. One of these abundant RNAs, ci-ankrd52, largely accumulates to its sites of transcription, associates with the elongation Pol II machinery, and acts as a positive regulator of Pol II transcription.4 Apparently also non-coding intronic segments of ciRNA transcripts can have a cis-regulatory role. CDR1-as stabilizes CDR1 mRNA expression, probably with a sense–antisense‐based feedback mechanism, where the antisense circRNA stimulates or stabilizes the sense mRNA with subsequent negative impact on antisense levels.31
The fairly well-characterized role of circRNAs to positively modulate host gene expression has foreseeable implications for stability of cell commitment choices such as in hematopoiesis.
In summary, circRNAs can regulate host gene expression but also participate to complex networks in which they compete for the binding of miRNAs (ceRNA networks) of RBPs or even for translation initiation. CircRNAs with different composition in terms of exon–intron inclusion result in a multitude of mechanisms that can affect transcriptome and proteome regulation. Through studies of specific tissues, cell types and conditions the evident versatility of circRNA is expected to reveal insight in all kinds of cellular processes.
CircRNAs in the hematopoietic compartment
RNA-seq analyses showed dynamic expression of circular isoforms independent of linear transcript dynamics from the same gene5 and cell- and differentiation stage-specific expression2, 13 prompting lines of research that focused on specific tissues. Knowing that circRNAs interfere in key cellular processes like self-renewal, proliferation and apoptosis there is growing interest to study of circRNA in the hematopoietic compartment. In the hematopoietic tissue, pioneering studies reported circRNA isoforms of key genes such as MLL25, 67 without receiving much resonance in the research community, probably due to the reported low expression of circular isoforms compared with abundant mRNAs encoding these key transcriptional regulators. In hindsight low expression of circRNAs of the above transcription factor can be understood from recent observations that in specific cell types highly expressed genes (in terms of expressed linear isoforms) give rise to relatively less circRNA compared with moderate- or low-expressed genes.5
CircRNA discovery by RNA-seq in hyperdiploid B-cell precursor-ALL and in sorted normal leukocyte cell populations
Aiming to discover new cancer-specific fusion transcripts in hyperdiploid B-lineage acute lymphoblastic leukemia, Salzman et al.11 exploited RNA-seq and found many transcripts with permutated exon order, which they called ‘scrambled exons’ and attributed to circularized RNAs. In five samples of hyperdiploid B-cell precursor-acute lymphoblastic leukemia they detected hundreds of circRNA transcripts with >700 circular isoforms comprising more than 10% of all transcript isoforms produced from a comparable number of genes. These circRNAs were however not a specific feature of the leukemic cells; PCR verified that scrambled exons were also detected in remission samples of the patients, in HeLa cells and in normal primary human cells. Also in sorted cells populations, naive B cells (CD19+), hematopoietic stem cells (CD34+) and neutrophils, circRNA isoforms expressed by >800 genes were identified, with circRNA expression accounting for >10% of gene expression. This study showed that a particular gene can produce circRNAs in more than one leukocyte type, but single replicate-based preliminary estimations suggest quantitative differences among cell types. This first indication that circRNAs are expressed both in normal and malignant hematopoietic cells informs the number of circRNAs in immature and lineage-specific blood cells but does not provide a more specific and useful interpretation of circRNA relevance for hematopoietic cell functions and pathology. For instance the study reported and validated a few most abundant transcripts with scrambled exons (ESYT2, FBXW4, CAMSAP1, KIAA0368, CLNS1A, FAM120A, MAP3K1, ZKSCAN1, MANBA, ZBTB46, NUP54, RARS and MGA) but did not pay particular attention to circRNAs from numerous genes that are important for normal hematopoiesis and present genomic aberrations or deregulated expression in leukemia.
According to published data11 we observed that circRNAs from genes related to B-cell differentiation and acute lymphoblastic leukemia (JAK2, PAX5, IKZF1, ETV6 and EBF1) are prevalently present in hyperdiploid leukemia compared with normal leukocytes samples. The latter is likely an underestimation, as only 54 genes were screened and circRNAs supported by only 1 read were not considered.
The same authors also investigated circRNA expression in 15 different cancer and non-cancer cell lines, detecting around 47 000 circle-specific splice junctions from 8500 genes. The validation of 8 candidates confirmed that all were true circRNAs.2 The study further specified that highly expressed circRNA showed cell-type-specific increase in expression that was not associated to an increase of the corresponding linear RNA. Notably, among others, the leukemia cell line K562 presented the largest number of genes (16 559) with evidence of circular RNA expression.
A reanalysis of the same data set using the CIRI method67 indicated that more universally shared circRNAs tend to have higher expression levels and verified that the expression patterns of linear transcripts of circRNA-encoding exons are more similar in cancer cells compared with non-cancer cell types, whereas cancer cells appear to have more diverse circRNA expression profiles, both considering exonic and intronic circRNAs.
Subsequently, a comparison of CD34+, CD19+, neutrophils and HEK293 (human embryonic kidney cells) considering only a single biological replicate per cell type (with sequencing depth around 20 million reads per samples) was reported.13 The study detected 1950 circRNAs of which 939 are exclusively expressed in CD19+ cells, 333 in CD34+, 194 in neutrophils and 60 in HEK293 cells. Nineteen circRNAs resulted to be shared between these cell types. The emphasis of this study was on the demonstration that circRNAs are in part cell-type-specific and are expressed in a developmental stage-related manner.
CircRNAs in whole-blood samples from healthy individuals
RNA-seq analysis of whole-blood samples68 showed that whole blood was very rich in circRNAs, comparable to the cerebellum, with consistent data comparing two biological replicates. Also in this study the emphasis laid on the numeric evaluation and the demonstration that circRNAs are a natural component of the transcriptome. Whole blood is composed of a gamma of different cell types. Moreover, the plasma component may also be a sink for circulating circRNA of non-hematopoietic cell origin and can be in fact explored for disease biomarkers, as previously proposed for solid tumors.9 We cannot exclude that plasma samples could also contain exogenous RNA. In malaria infection, thousands of very short circRNAs are produced by Plasmodium falciparum,69 including dozens of circRNAs harboring >100 binding sites for a given human miRNA, pointing to highly versatile parasite–host interactions. Similarly, as already proposed for virus–host interactions, circRNAs of viral origin might sponge host miRNAs and vice versa.70
CircRNAs in platelets
Alhasan et al.71 reported circRNA enrichment in platelets that they ascribed mainly to differential decay of linear RNA, considering the particular circRNA resistance to degradation. In the past integration of transcriptome and proteome data of platelets had given somewhat conflicting results.72 Extensive degradation of linear RNA isoforms leaving circRNAs intact, which results in an extensive reduction of the translatable RNAs provides now a straightforward explanation for this apparent disparity. This study demonstrated that circRNAs are highly enriched not only in platelets but also in erythrocytes relative to nucleated cells finding that >3000 genes show 17- to 188-fold relative enrichment of circRNAs.
Fusion-circRNAs derived from chromosomal translocations have oncogenic role
Fusion-circRNA discovery
Very recently fusion-circRNAs (f-circRNAs) derived from transcribed exons of chimeric genes generated by cancer-associated chromosomal translocation were discovered and proven to be oncogenic according to in vitro and in vivo experiments73 (Figure 5). Instead of a discovery-driven approach this study used informed guessing to directly detect transcript circularization around the breakpoint/fusion region of two well-known recurrent leukemia-related translocations. The authors hypothesized that juxtaposition of complementary sequences in introns at either side of the fusion regions could favor the formation of circRNAs and searched specifically for circRNAs expressed from fusion genes. f-circRNAs were thus detected by RT–PCR and then confirmed using RNA-seq and custom bioinformatics procedures, in promyelocytic leukemia with a PML/RARα and acute myeloid leukemia with an MLL/AF9 fusion (Figure 5a). Both translocations gave rise to more than one f-circRNA characterized by different backsplice junctions, both in patient samples and in patient-derived cell lines. The discovery was also extended to solid tumors showing f-circRNAs transcription in translocated Ewing sarcoma and lung cancer.

f-circRNAs derived from chromosomal translocations have oncogenic role. (a) Transcription of fusion genes generated by cancer-associated chromosomal translocation could generate both linear mRNA coding for oncogenic fusion proteins and f-circRNAs.73 The figure depicts the example of f-CircM9_1 expressed in cells harboring the well-known acute myeloid leukemia MLL/AF9 t(9;11)) translocation: f-CircM9_1 includes two sequences not present in the normal genome, the MLL exon 8 and AF9 exon 6 fusion junction derived from the chromosomal translocation, and the backsplice junction connecting MLL exon 7 with AF9 exon 6; (b) f-CircM9 was demonstrated to be proto-oncogenic in vitro (increasing proliferation rate and foci forming ability in mouse embryonic fibroblasts, MEF), and required for leukemic cell (THP1) viability. f-CircM9 alone resulted not sufficient to trigger leukemia in vivo when expressed in HSC xenografted in mice. Concurrent expression of f-circM9 and MLL/AF9 fusion protein contributed to leukemia progression in vivo and ex vivo cells expressing f-circM9 and MLL/AF9 displayed increased ability to proliferate and to form colonies. Furthermore, f-circM9 expression in MLL/AF9 mouse model cells increased the resistance to leukemia treatments suggesting that f-circM9 impacts to pre-clinical therapeutic outcome.
f-circRNAs are proto-oncogenic and a requisite for leukemic cells viability
Guarnerio et al.73 showed that f-circRNAs (f-circPR and f-circM9) expression in leukemic cells increases cell proliferation and clonogenicity and that f-circRNA silencing reverted the phenotype, demonstrating that these f-circRNAs are biologically active and exert pro-proliferative and proto-oncogenic activities(Figure 5b). Moreover, shRNA-based knockdown of f-circM9 in leukemic THP1 cells resulted in increased apoptosis showing that f-circRNAs have an important role in maintaining the viability of leukemic cells.
In vivo study of f-circRNAs
Human leukemic cells in vivo expressing f-circRNAs sustain disease progression in mouse. On the other hand, f-circRNAs alone did not trigger leukemia. Cells expressing f-circM9 together with the MLL/AF9 fusion protein have an increased ability to proliferate and form colonies than cells expressing the fusion protein alone, strengthening the hypothesis that f-circRNAs contribute to the leukemogenic process in vivo (Figure 5b). Human leukemic cells in vivo expressing f-circRNAs sustain disease progression in mouse. Moreover, f-circRNAs expression could provide tumor cells protection to standard leukemia treatment, as arsenic trioxide, and confer survival advantage to leukemic cells in response in addition to standard-of-care leukemia treatment with cytarabine. Thus, according to these experiments in a pre-clinical setting f-circM9 could impact therapeutic outcomes.
Interestingly, Guarnerio et al.73 argued that the latency of leukemia development in animal models could be due to the absence of f-circRNAs in modeled expression of intron-less fusion genes. Even if it does provide neither data nor hypotheses on the mechanisms underlying the observed pathogenetic effect of f-circRNA, the study73 is a breakthrough for leukemia research. In addition, as noticed by the authors, not only the expression of f-circRNA but also the reduction of circRNAs expressed from the non-translocated allele partner genes can contribute to the pathogenic effect.
Conclusions and outlook
CircRNA expression further challenges a simplistic definition of ‘gene’, reinforcing the concept that genes are complex transcriptional units, and that the sequence of a given genomic region is a sort of palimpsest (ancient parchment on which the original text was overwritten multiple times) that contains multiple, interleaved and overlapping information parcels.74 Transcripts from the same locus use a common sequence in different ways, and perform distinct biological roles. In addition, circRNAs add new hints to our understanding of the alternative use and reuse of RNA sequences to produce different products and even small RNAs, as known in the case of miRtrons,75 of tRNA- or snoRNA-derived miRNAs74, 76 and moRNAs,77, 78, 79 that are expressed in blood cells,78, 79 and were shown to have pathogenic relevance in B-cell lymphomas.80 Ultimately gene expression studies need to disentangle the expression of linear and circRNAs expressed from each locus in order to dissect the distinct or complementary processes in which they participate.
A large body of data regarding molecular circuits that control cellular differentiation of the hematopoietic system is available and its deregulation in malignancies in association with genomic lesions is increasingly understood. In hematopoiesis, differentiated cell states are controlled by densely interconnected transcriptional circuits81 in a seemingly hierarchical process of binary fate decisions, but the stiffness of cell fate may be more fluid82 allowing for epigenetic regulation in response to mature blood cell demand. We envisage that circRNAs studies of hematopoietic cell stages will further elucidate how cell fate fluidity may depend on stably present circRNAs of key cell stage mRNAs.
Gene expression profiling of the protein-coding transcriptome has been very useful for the study of hematopoietic malignancies but will become more complete when integrated with circRNA expression data. Among other things it can be expected to elucidate discordant gene–protein expression often revealed for marker proteins of hematopoietic cell stages (for example, CD10, CD22 and CD38). Further, induced pluripotent stem cell modeling of hematopoietic diseases may also benefit from circRNA studies, hitherto nothing is known about the behavior of cricRNAs in reprogramming procedures of induced pluripotent stem cell generation.
Recent RNA-seq data outlined transcriptional diversity in terms of (linear) alternative isoform-ratio variations among hematopoietic cells47, 83 and of non-coding RNA’s impact in hematopoietic lineage differentiation.84 Specific miRNAs are expressed in a developmental-stage-specific manner.85, 86 miRNAs and other small RNAs are differentially expressed in disease.79
As in last years we abandoned the concept of the centrality of coding fraction of the transcriptome, the discovery of circRNAs made clear that the study of the linear RNAs only provides an incomplete picture of the cellular complexity. Focusing on linear transcription only we miss important elements, both for data interpretation and experimental design.
Today we have an increased appreciation of circRNA abundance, evolutionary conservation and diversity of functions and interactions. Specific data emerged of high and regulated circRNA expression in normal and malignant blood cells. The recent discovery and functional study of f-circRNAs provided important clues of the oncogenic role of this aberrant circRNA in leukemogenesis and of their relevance in modulating therapeutic outcome. Together these data clearly indicate that further studies of circular isoforms from different cell types and stages of the hematopoietic compartment and by rearranged or mutated genomes are warranted to better estimate the position of these new regulators in hematopoietic cell development and derived malignancies. The route toward elucidating circRNA biology is still long. Even a consistent nomenclature for circRNAs is sorely missed.
Definitely, circRNAs and their diverse molecular interactions participate to the circuitries that regulate the final cellular protein output adding to the richness and complexity of the underlying mechanisms. The particular stability of circRNAs may also make them valuable disease markers that can be identified in various body fluids and we envisage that a better understanding of circRNA biology will inform innovative therapeutic targets.
References
Lasda E, Parker R . Circular RNAs: diversity of form and function. RNA 2014; 20: 1829–1842.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO . Cell-type specific features of circular RNA expression. PLoS Genet 2013; 9: e1003777.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 2015; 22: 256–264.
Zhang Y, Zhang X-O, Chen T, Xiang J-F, Yin Q-F, Xing Y-H et al. Circular intronic long noncoding RNAs. Mol Cell 2013; 51: 792–806.
Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell 2015; 58: 870–885.
Dean M, Fojo T, Bates S . Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5: 275–284.
Costa D, Gigoni A, Würth R, Cancedda R, Florio T, Pagano A . Metformin inhibition of neuroblastoma cell proliferation is differently modulated by cell differentiation induced by retinoic acid or overexpression of NDM29 non-coding RNA. Cancer Cell Int 2014; 14: 59.
Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T et al. Correlation of circular RNA abundance with proliferation-exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 2015; 5: 8057.
Li P, Chen S, Chen H, Mo X, Li T, Shao Y et al. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta 2015; 444: 132–136.
Bahn JH, Zhang Q, Li F, Chan T-M, Lin X, Kim Y et al. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem 2015; 61: 221–230.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO . Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012; 7: e30733.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013; 19: 141–157.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013; 495: 333–338.
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P et al. Genome-wide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 2014; 9: 1966–1980.
Zhang Z, Qi S, Tang N, Zhang X, Chen S, Zhu P et al. Discovery of replicating circular RNAs by RNA-seq and computational algorithms. PLoS Pathog 2014; 10: e1004553.
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 2015; 10: 170–177.
Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang J-L et al. The Biogenesis of Nascent Circular RNAs. Cell Rep 2016; 15: 611–624.
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK . Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci USA 1976; 73: 3852–3856.
Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert J, Oliner JD et al. Scrambled exons. Cell 1991; 64: 607–613.
Cocquerelle C, Mascrez B, Hetuin D, Bailleul B . Mis-splicing yields circular RNA molecules. FASEB J 1993; 7: 155–160.
Chao CW, Chan DC, Kuo A, Leder P . The mouse formin (Fmn) gene: abundant circular RNA transcripts and gene-targeted deletion analysis. Mol Med 1998; 4: 614.
Pasman Z, Been M, Garcia-Blanco M . Exon circularization in mammalian nuclear extracts. RNA 1996; 2: 603.
Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993; 73: 1019–1030.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495: 384–388.
Caldas C, So CW, MacGregor A, Ford AM, McDonald B, Chan LC et al. Exon scrambling of MLL transcripts occur commonly and mimic partial genomic duplication of the gene. Gene 1998; 208: 167–176.
Bailleul B . During in vivo maturation of eukaryotic nuclear mRNA, splicing yields excised exon circles. Nat Struct Mol Biol 1996; 24: 1015–1019.
Zaphiropoulos PG . Exon skipping and circular RNA formation in transcripts of the human cytochrome P-450 2C18 gene in epidermis and of the rat androgen binding protein gene in testis. Mol Cell Biol 1997; 17: 2985–2993.
Li X-F, Lytton J . A circularized sodium-calcium exchanger exon 2 transcript. J Biol Chem 1999; 274: 8153–8160.
Surono A, Takeshima Y, Wibawa T, Ikezawa M, Nonaka I, Matsuo M . Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum Mol Genet 1999; 8: 493–500.
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE . Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 2010; 6: e1001233.
Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ et al. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 2011; 30: 4414–4422.
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014; 56: 55–66.
Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung L-H et al. Exon circularization requires canonical splice signals. Cell Rep 2015; 10: 103–111.
Wang Y, Wang Z . Efficient backsplicing produces translatable circular mRNAs. RNA 2015; 21: 172–179.
Kelly S, Greenman C, Cook PR, Papantonis A . Exon skipping is correlated with exon circularization. J Mol Biol 2015; 427: 2414–2417.
Dolci S, Grimaldi P, Geremia R, Pesce M, Rossi P . Identification of a promoter region generating Sry circular transcripts both in germ cells from male adult mice and in male mouse embryonal gonads. Biol Reprod 1997; 57: 1128–1135.
Salzman J . Circular RNA expression: its potential regulation and function. Trends Genet 2016; 32: 309–316.
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA et al. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015; 160: 1125–1134.
Lee Y, Rio DC . Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 2015; 84: 291–323.
Liu S, Cheng C . Alternative RNA splicing and cancer. Wiley Interdiscip Rev RNA 2013; 4: 547–566.
Bisognin A, Pizzini S, Perilli L, Esposito G, Mocellin S, Nitti D et al. An integrative framework identifies alternative splicing events in colorectal cancer development. Mol Oncol 2014; 8: 129–141.
Yoshida K, Ogawa S . Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 2014; 5: 445–459.
Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 2011; 118: 6904–6908.
Martinez-Avilés L, Besses C, Álvarez-Larrán A, Camacho L, Pairet S, Fernández-Rodriguez C et al. Mutations in the RNA splicing machinery genes in myelofibrotic transformation of essential thrombocythaemia and polycythaemia vera. Br J Haematol 2014; 164: 605–607.
Tefferi A, Lasho T, Finke C, Knudson R, Ketterling R, Hanson C et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia 2014; 28: 1472–1477.
Hou H-A, Liu C-Y, Kuo Y-Y, Chou W-C, Tsai C-H, Lin C-C et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget 2016; 7: 9084–9101.
Chen L, Kostadima M, Martens JH, Canu G, Garcia SP, Turro E et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science 2014; 345: 1251033.
Makino DL, Halbach F, Conti E . The RNA exosome and proteasome: common principles of degradation control. Nat Rev Mol Cell Biol 2013; 14: 654–660.
Chen L-L, Yang L . Regulation of circRNA biogenesis. RNA Biol 2015; 12: 381–388.
Thomas LF, Sætrom P . Circular RNAs are depleted of polymorphisms at microRNA binding sites. Bioinformatics 2014; 30: 2243–2246.
Bretscher MS . Translocation in protein synthesis: a hybrid structure model. Nature 1968; 218: 675–677.
Chen C-Y, Sarnow P . Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 1995; 268: 415.
Abe N, Matsumoto K, Nishihara M, Nakano Y, Shibata A, Maruyama H et al. Rolling circle translation of circular RNA in living human cells. Sci Rep 2015; 5: 16435.
AbouHaidar MG, Venkataraman S, Golshani A, Liu B, Ahmad T . Novel coding, translation, and gene expression of a replicating covalently closed circular RNA of 220 nt. Proc Natl Acad Sci USA 2014; 111: 14542–14547.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 2015; 18: 603–610.
Jeck WR, Sharpless NE . Detecting and characterizing circular RNAs. Nat Biotechnol 2014; 32: 453.
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R et al. Circular RNA: a new star of noncoding RNAs. Cancer Lett 2015; 365: 141–148.
Li F, Zhang L, Li W, Deng J, Zheng J, An M et al. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/β-catenin pathway. Oncotarget 2015; 6: 6001–6013.
Huang G, Zhu H, Shi Y, Wu W, Cai H, Chen X . cir-ITCH plays an inhibitory role in colorectal cancer by regulating the Wnt/β-catenin pathway. PLoS One 2015; 10: e0131225.
Xie H, Ren X, Xin S, Lan X, Lu G, Ss Y et al. Emerging roles of circRNA001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget 2016; 7: 26680–26691.
Wang K, Long B, Liu F, Wang J-X, Liu C-Y, Zhao B et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J 2016; 37: 2602–2611.
Ala U, Karreth FA, Bosia C, Pagnani A, Taulli R, Léopold V et al. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc Natl Acad Sci USA 2013; 110: 7154–7159.
Guo JU, Agarwal V, Guo H, Bartel DP . Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014; 15: 409.
Thomson DW, Dinger ME . Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 2016; 17: 272–283.
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB . Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nat Struct Mol Biol 2016; 44: 2846–2858.
Zuna J, Burjanivova T, Mejstrikova E, Zemanova Z, Muzikova K, Meyer C et al. Covert preleukemia driven by MLL gene fusion. Genes Chromosomes Cancer 2009; 48: 98–107.
Gao Y, Wang J, Zhao F . CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 2015; 16: 4.
Memczak S, Papavasileiou P, Peters O, Rajewsky N . Identification and characterization of circular RNAs as a new class of putative biomarkers in human blood. PloS one 2015; 10: e0141214.
Broadbent KM, Broadbent JC, Ribacke U, Wirth D, Rinn JL, Sabeti PC . Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genomics 2015; 16: 454.
Ghosal S, Das S, Sen R, Basak P, Chakrabarti J . Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet 2013; 4: 283.
Alhasan AA, Izuogu OG, Al-Balool HH, Steyn JS, Evans A, Colzani M et al. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood 2016; 127: e1–e11.
Londin ER, Hatzimichael E, Loher P, Edelstein L, Shaw C, Delgrosso K et al. The human platelet: strong transcriptome correlations among individuals associate weakly with the platelet proteome. Biol Direct 2014; 9: 3.
Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM et al. Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell 2016; 165: 289–302.
Tuck AC, Tollervey D . RNA in pieces. Trends Genet 2011; 27: 422–432.
Schamberger A, Sarkadi B, Orbán TI . Human mirtrons can express functional microRNAs simultaneously from both arms in a flanking exon-independent manner. RNA Biol 2012; 9: 1177–1185.
Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R . Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor-independent, dicer-dependent small RNAs. Genes Dev 2008; 22: 2773–2785.
Bortoluzzi S, Biasiolo M, Bisognin A . MicroRNA–offset RNAs (moRNAs): by-product spectators or functional players? Trends Mol Med 2011; 17: 473–474.
Bortoluzzi S, Bisognin A, Biasiolo M, Guglielmelli P, Biamonte F, Norfo R et al. Characterization and discovery of novel miRNAs and moRNAs in JAK2V617F-mutated SET2 cells. Blood 2012; 119: e120–e130.
Guglielmelli P, Bisognin A, Saccoman C, Mannarelli C, Coppe A, Vannucchi AM et al. Small RNA sequencing uncovers new miRNAs and moRNAs differentially expressed in normal and primary myelofibrosis CD34+ cells. PloS One 2015; 10: e0140445.
Maute RL, Schneider C, Sumazin P, Holmes A, Califano A, Basso K et al. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci USA 2013; 110: 1404–1409.
Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011; 144: 296–309.
Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016; 351: aab2116.
Shi L, Lin Y-H, Sierant MC, Zhu F, Cui S, Guan Y et al. Developmental transcriptome analysis of human erythropoiesis. Hum Mol Genet 2014; 23: 4528–4542.
Morlando M, Ballarino M, Fatica A . Long non-coding RNAs: new players in hematopoiesis and leukemia. Front Med (Lausanne) 2015; 2: 23.
O’Connell RM, Chaudhuri AA, Rao DS, Gibson WSJ, Balazs AB, Baltimore D . MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc Natl Acad Sci USA 2010; 107: 14235–14240.
Zhang L, Sankaran VG, Lodish HF . MicroRNAs in erythroid and megakaryocytic differentiation and megakaryocyte-erythroid progenitor lineage commitment. Leukemia 2012; 26: 2310–2316.
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 2010; 28: 2529–2537.
Otto C, Stadler PF, Hoffmann S . Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinformatics 2014; 30: 1837–1843.
Song X, Zhang N, Han P, Moon B-S, Lai RK, Wang K et al. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res 2016; 44: e87.
Chuang T-J, Wu C-S, Chen C-Y, Hung L-Y, Chiang T-W, Yang M-Y . NCLscan: accurate identification of non-co-linear transcripts (fusion, trans -splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res 2015; 44: e29.
Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nat Struct Mol Biol 2010; 38: e178.
Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C et al. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol 2015; 16: 126.
Izuogu OG, Alhasan AA, Alafghani HM, Santibanez-Koref M, Elliott DJ, Jackson MS . PTESFinder: a computational method to identify post-transcriptional exon shuffling (PTES) events. BMC Bioinformatics 2016; 17: 1–11.
Hansen TB, Venø MT, Damgaard CK, Kjems J . Comparison of circular RNA prediction tools. Nucleic Acids Res 2015; 44: e58.
Glažar P, Papavasileiou P, Rajewsky N . circBase: a database for circular RNAs. RNA 2014; 20: 1666–1670.
Li J-H, Liu S, Zhou H, Qu L-H, Yang J-H . starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nat Struct Mol Biol 2013; 42: D92–D97.
Liu Y-C, Li J-R, Sun C-H, Andrews E, Chao R-F, Lin F-M et al. CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res 2015; 44: D209–D215.
Acknowledgements
We thank for financial support to SB Fondazione Cassa di Risparmio di Padova e Rovigo Progetti di Eccellenza 2011/2012 by Ministero dell’Istruzione, dell’Università e della Ricerca PRIN 2010/11 (2010NYKNS7_002) and University of Padova; to GteK Fondazione Cariplo, Ministero dell’Istruzione, dell’Università e della Ricerca FIRB RBAP11TF7Z and University of Padova. AB is recipient of a PhD fellowship from the Fondazione Città della Speranza. Silvia Bresolin, Luca Trentin, Andrea Bisognin, Giulia Anselmi, Elena Boldrin and Lueder H Meyer are acknowledged for fruitful discussions.
Author contributions
Study design and concept: all authors; bibliographic review: AB, GteK and SB; bioinformatic method review: AB and EG; figure concepts and drawing: AB and SB; manuscript writing: GteK and SB; critical review and approval of the final manuscript: all authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bonizzato, A., Gaffo, E., te Kronnie, G. et al. CircRNAs in hematopoiesis and hematological malignancies. Blood Cancer Journal 6, e483 (2016). https://doi.org/10.1038/bcj.2016.81
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2016.81
This article is cited by
-
CircFBXW7 in patients with T-cell ALL: depletion sustains MYC and NOTCH activation and leukemia cell viability
Experimental Hematology & Oncology (2023)
-
Roles of circRNAs in hematological malignancies
Biomarker Research (2022)
-
Analysis of circRNAs and circRNA-associated competing endogenous RNA networks in β-thalassemia
Scientific Reports (2022)
-
Global identification of circular RNAs in imatinib (IM) resistance of chronic myeloid leukemia (CML) by modulating signaling pathways of circ_0080145/miR-203/ABL1 and circ 0051886/miR-637/ABL1
Molecular Medicine (2021)
-
Novel circRNA discovery in sheep shows evidence of high backsplice junction conservation
Scientific Reports (2021)
