
Abstract
Chronic myelomonocytic leukemia (CMML) is a clonal stem cell disorder associated with peripheral blood monocytosis and an inherent tendency to transform to acute myeloid leukemia. CMML has overlapping features of myelodysplastic syndromes and myeloproliferative neoplasms. Clonal cytogenetic changes are seen in ~30%, whereas gene mutations are seen in >90% of patients. Common cytogenetic abnormalities include; trisomy 8, -Y, -7/del(7q), trisomy 21 and del(20q), with the Mayo–French risk stratification effectively risk stratifying patients based on cytogenetic abnormalities. Gene mutations frequently involve epigenetic regulators (TET2 ~60%), modulators of chromatin (ASXL1 ~40%), spliceosome components (SRSF2 ~50%), transcription factors (RUNX1 ~15%) and signal pathways (RAS ~30%, CBL ~15%). Of these, thus far, only nonsense and frameshift ASXL1 mutations have been shown to negatively impact overall survival. This has resulted in the development of contemporary, molecularly integrated (inclusive of ASXL1 mutations) CMML prognostic models, including Molecular Mayo Model and the Groupe Français des Myélodysplasies model. Better understanding of the prevalent genetic and epigenetic dysregulation has resulted in emerging targeted treatment options for some patients. The development of an integrated (cytogenetic and molecular) prognostic model along with CMML-specific response assessment criteria are much needed future goals.
Similar content being viewed by others

Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal stem cell disorder with overlapping features of myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN).1, 2 CMML often results in peripheral blood monocytosis and has an inherent tendency to transform to acute myeloid leukemia (AML; ~30%).2, 3 Clonal cytogenetic changes are seen in ~30% of patients,4, 5 whereas molecular and epigenetic abnormalities are seen in >90%.6, 7 CMML is further subclassified into CMML-1 (<5% circulating blasts and <10% bone marrow (BM) blasts) and CMML-2 (5–19% circulating blasts, 10–19% BM blasts or when Auer rods are present irrespective of the blast count),6, 8, 9 with approximate median overall survival (OS) of 38 and 24 months, respectively.6, 7
Gene mutations in CMML involve epigenetic regulators (TET2 ~60%), chromatin/histone modulators (ASXL1 ~40%), spliceosome components (SRSF2 ~50%), transcription factors (RUNX1 ~15%) and cell signaling (RAS ~30%, CBL ~15%).2, 6, 7, 10 Among these, thus far, on multivariable analyses that have included additional CMML relevant factors, only ASXL1 mutations (frameshift and nonsense) have been shown to be prognostically detrimental.6, 7 This has led to the incorporation of ASXL1 mutations into molecular prognostic models, such as the Molecular Mayo Model and the Groupe Francais des Myelodysplasies (GFM) model.6, 7 In the current review, we discuss and summarize the prevalence, phenotypic, prognostic and therapeutic impact of cytogenetic and molecular abnormalities in CMML.
Cytogenetic abnormalities in CMML
The 2008 World Health Organization (WHO) criteria define CMML as a disorder characterized by: (a) persistent peripheral blood monocytosis >1 × 109/l, (b) absence of the Philadelphia chromosome and the BCR-ABL1 fusion oncogene, (c) absence of the PDGFRA or PDGFRB gene rearrangements, (d) <20% blasts and promonocytes in the peripheral blood and BM, and (e) dysplasia involving one or more myeloid lineages.1 If myelodysplasia is absent or minimal, the diagnosis of CMML can still be made if the other requirements are met and an acquired, clonal or molecular genetic abnormality is present in the hematopoietic cells or if the monocytosis has persisted for at least 3 months and other causes of monocytosis have been excluded.1, 2
The BCR-ABL1 fusion oncogene defines chronic myeloid leukemia, a unique myeloid neoplasm in which monocytosis is uncommon.11 The platelet-derived growth factor receptors alpha and beta (PDGFRA—chromosome 4q12 and PDGFRB—chromosome 5q31-q32) are type III receptor tyrosine kinases. Chromosomal translocations involving PDGFRA/B have been associated with myeloid neoplasms characterized by prominent eosinophilia and responsiveness to imatinib.12, 13 At times, PDGFR-rearranged myeloid neoplasms can be associated with monocytosis and BM dysplasia, but given their unique responsiveness to imatinib, these are no longer classified as CMML. Patients presenting with a clinical phenotype of CMML with eosinophilia should be assessed for the t(5;12)(q31-q32;p13), giving rise to the ETV6(TEL)-PDGFRB fusion oncogene.14 The association between monocytosis and PDGFRA rearrangements is uncommon.15
Clonal cytogenetic abnormalities are seen in ~30% of CMML patients.5, 8, 16, 17 Common alterations include: trisomy 8 (+8), -Y, abnormalities of chromosome 7 (monosomy 7 and del7q), trisomy 21 (+21), and complex karyotypes (Table 1).5 Unlike in MDS, sole del(5q) (<1%) and monosomal karyotypes (~10%) are infrequent.4, 18, 19 Based on these findings, the Spanish cytogenetic risk stratification system was developed, categorizing patients into three groups; high risk (trisomy 8, chromosome 7 abnormalities or complex karyotype), intermediate risk (all chromosomal abnormalities, except for those in the high- and low-risk categories), and low risk (normal karyotype or -Y), with 5-year OS of 4, 26 and 35%, respectively.5
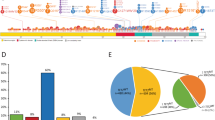
Recently, in a large international collaborative study, 409 patients with CMML were analyzed for cytogenetic and molecular abnormalities (268 (66%) and 141 (34%) from the Mayo Clinic and French Consortium, respectively).4 Thirty percent displayed an abnormal karyotype, with common abnormalities being +8 (23%), -Y (20%), −7/7q-(14%), 20q- (8%), +21 (8%) and der(3q) (8%).4 A step-wise survival analysis resulted in three distinct cytogenetic risk categories: high (complex and monosomal karyotypes), intermediate (all abnormalities not in the high- or low-risk groups) and low (normal, sole -Y and sole der(3q)), with median survivals of 3 (hazard ratio (HR) 8.1, 95% confidence interval (CI) 4.6–14.2), 21 (HR 1.7, 95% CI 1.2–2.3) and 41 months, respectively (Figure 1).4 In multivariable analysis, this particular cytogenetic risk stratification remained significant in the context of the Molecular Mayo Model (P< 0.0001), MD Anderson prognostic model (P<0.0001) and the GFM model (P<0.0001) and was effective in predicting leukemic transformation (P=0.004).4

OS of 409 patients with WHO-defined CMML, stratified by the Mayo–French cytogenetic risk stratification system.
Molecular and epigenetic abnormalities in CMML
Gene mutations are seen in >90% of patients with CMML (Figure 2).20, 21, 22 These abnormalities can be broadly classified into the following categories:
-
1
Mutations involving epigenetic regulator genes: TET2 (~60%), DNMT3A, IDH1, and IDH2.
-
2
Mutations involving chromatin regulation and histone modification: ASXL1 (~40%), EZH2, UTX, EED, and SUZ12.
-
3
Mutations involving the splicing machinery (pre-mRNA splicing): SF3B1, SRSF2 (~50%), U2AF1, and ZRSR2.
-
4
Mutations involving the cohesin complex: STAG2, BCOR, SMC3, SMC1A, and RAD21.
-
5
Mutations involving DNA damage response genes: Tp53 (~1%) and PHF6.
-
6
Mutations in signal transduction and cellular/receptor tyrosine kinase pathways: JAK2, SH2B3 (LNK), KRAS, NRAS (RAS ~30%), CBL (~10–15%), FLT3, and NPM1
-
7
Others: RUNX1 (transcription factor) and SETBP1 (~15%).

Spectrum of gene mutations seen in patients with CMML.
The genetic heterogeneity of CMML, in patients and in between patients, suggests that the disease has different potential evolutional trajectories.21, 23 Current studies suggest that the preferred order of mutational accumulation is TET2 (or IDH1/2) or ASXL1 (EZH2) first, spliceosome component mutations (SRSF2, SF3B1, U2AF1 or ZRSR2) next, followed by transcription factor mutations (RUNX1) and then signal pathway gene mutations (RAS, CBL), inducing GM-CSF (granulocyte macrophage-colony stimulating factor) hypersensitivity and myeloproliferation (Figure 3).21, 23, 24

Mutational evolution and clonal hierarchy in patients with CMML.
Mutations in epigenetic regulator genes impacting DNA methylation and hydroxy-methylation (TET2, DNMT3A, IDH1 and IDH2)
TET2 (ten-eleven translocation (TET) oncogene family member 2—chromosome 4q24) is a member of the TET family of proteins (TET1–TET3).25 TET2 mutations are seen in ~15% of myeloid neoplasms,26 with individual mutational frequencies of; ~60%—CMML, ~15%—MDS, ~15%—polycythemia vera and primary myelofibrosis (PMF), ~20%—secondary AML and ~30%—systemic mastocytosis, with limited prognostic significance.7, 26, 27, 28 TET2 has a dioxygenase enzymatic activity and catalyzes the conversion of 5-methyl-cytosine (5-mc) to 5-hydroxy-methyl-cytosine (5-hmc). 5-hmC represents a new base in genomic DNA, which may have a specific effect on transcription and/or may represent an intermediate process in DNA demethylation.29, 30 5-hmC is most often found at transcription start sites and within gene bodies (preferentially in gene exons).31
Ko et al.32 reported that loss of 5-mC was a remarkable characteristic in CMML patients with TET2 mutations and found 2510 differentially hypomethylated regions and only two hypermethylated regions. In contrast, Figueroa et al.33 studied TET2 mutant leukemic cells and identified a hypermethylation phenotype, including 129 differentially methylated regions. Yamazaki et al.25 using bisulfite pyrosequencing, confirmed that TET2 mutations affect global methylation in CMML but hypothesized that most of the changes were likely to be outside gene-promoter regions.
In four mouse models, the deletion of TET2 has resulted in progressive expansion of the hematopoietic progenitor compartment, increased hematopoietic stem cell self-renewal and the progressive development of a proliferative myeloid malignancy, similar to CMML.34, 35, 36
Although TET2 mutations are widely prevalent in CMML (~60%), thus far, they have not been shown to independently impact either OS or leukemia-free survival (LFS).7 In a recent study, TET2 mutations were seen in 46% of CMML patients and the absence of TET2 mutations negatively impacted OS. Additionally, the presence of clonal TET2 mutations, in the absence of clonal ASXL1 mutations (ASXL1wt/TET2mut), had a favorable impact on OS.37 The mechanism behind this association is unclear. In MDS and younger patients with CMML (age <65 years), the presence of clonal TET2 mutations, in the absence of clonal ASXL1 mutations, has been associated with a favorable response to hypomethylating agents (5–azacitidine and decitabine).38, 39 Treatment data on this study cohort was incomplete.37
Mutations involving IDH1 (isocitrate dehydrogenase—chromosome 2q34) and IDH2 (chromosome 15q26.1) are uncommon in CMML (<5%) and are mutually exclusive with TET2 mutations.7, 40 IDH1/2 normally participates in the citric acid cycle and converts isocitrate to 5-alpha-ketoglutarate (Figure 4). IDH mutations confer a new enzymatic function to these enzymes, resulting in the development of an onco metabolite termed 2-hydroxyglutarate (2-HG).29 2-HG in turn impairs other enzymes, including TET2 and JMJC (Jumonji-C domain containing) family of histone lysine demethylases,41 contributing to leukemogenesis.

Isocitrate dehydrogenase mutations in CMML.
DNMT3A (DNA methyltransferase 3A—chromosome 2p23.3) mutations are seen in MDS (~10%),42 cytogenetically normal AML (~30%),29 PMF (~10%)43 and CMML (<5%).7 In CMML, thus far, they have not been shown to impact either OS or LFS.7
Mutations in epigenetic regulator genes impacting chromatin and histone modification (ASXL1, EZH2, EED, SUZ12 and UTX)
The ASXL1 (additional sex combs-like 1—chromosome 20q11) gene regulates chromatin by interacting with the polycomb group repressive complex proteins (PRC1 and PRC2).44 The PRC2 contains histone 3 lysine 27 (H3K27) methyltransferase, EZH2 (enhancer of zeste homolog 2) and its partners EED (embryonic ectoderm development) and SUZ12 (suppressor of zeste 12 homolog) and produces the H3K27 trimethyl mark (Figure 5).45 Histone 2A lysine 119 (H2AK119Ub) and H3K27me3 have synergistic roles in PRC-mediated gene repression.45, 46 In a seminal paper, Abdel-Wahab et al.46 demonstrated that ASXL1 mutations resulted in loss of PRC2-mediated H3K27 trimethylation. In addition, Balasubramani et al.45 demonstrated that ASXL1 truncations conferred enhanced activity on the ASXL1-BAP1 (BRCA-associated protein 1) complex. This resulted in global erasure of H2AK119Ub and depletion of H3K27me3, promoting dysregulated transcription.

ASXL1 and EZH2 mutations in CMML.
ASXL1 mutations are common in myeloid neoplasms, including MDS,44, 47 CMML,7, 9, 48 PMF44, 49 and AML,47, 50 with respective mutational frequencies ranging from 15 to 20, 40–50, 20–35 and 5–10%.20 In general, they are associated with an aggressive phenotype.48, 49, 50 In MDS, Bejar et al.51 identified ASXL1 mutations in 63 (14.4%) of 439 MDS patients and found these to be IPSS (International Prognostic Scoring System) independent predictors for shortened OS. In a large (879 patients) PMF collaborative study, ASXL1 mutations were identified in 20% of patients and were associated with older age, presence of constitutional symptoms, leukocytosis and circulating blasts.52 In systemic mastocytosis, ASXL1 mutations were seen in 9 (14%) of 62 patients and predicted for a shortened OS.53 In AML, ASXL1 mutations have been found to be mutually exclusive with the favorable NPM1 mutations, with some,54, 55 but not all,56 studies demonstrating an independent prognostic impact.
In CMML, ~40% of patients carry ASXL1 mutations, with the most frequent being the c.1934dupG; p.G646WfsX12 (~50%).7, 9 Although initially some investigators had considered c.1934dupG; p.G646WfsX12 to be a PCR artefact,57 subsequent studies have demonstrated its absence in germ-line DNA and control DNA, establishing it to be a bona fide mutation.20, 58 In CMML, ASXL1 mutations are associated with a proliferative phenotype, including higher WBC (white blood counts), higher absolute monocyte count (AMC) and the presence of circulating immature myeloid cells (IMC).7, 9, 20
The discovery of gene mutations in CMML has led to their incorporation into prognostic models. A Mayo Clinic study (n=226) analyzed several parameters, including ASXL1 mutations; on multivariable analysis, risk factors for survival included HB (hemoglobulin) <10 gm/dl, platelet count <100 × 109/l, AMC>10 × 109/l and circulating IMC.9 ASXL1 mutations did not impact either the OS or the LFS. The study resulted in the development of the Mayo prognostic model, with three risk categories, low (0 risk factor), intermediate (1 risk factor) and high (⩾2 risk factors), with median survivals of 32, 18.5 and 10 months, respectively.9 The GFM demonstrated an adverse prognostic effect for ASXL1 mutations in 312 patients with CMML; additional risk factors on multivariable analysis included age >65 years, WBC>15 × 109/l, platelet count <100 × 109/l and HB<10 gm/dl in females and <11 gm/dl in males.7 The GFM model assigns three adverse points for WBC>15 × 109/l and two adverse points for each one of the remaining risk factors, resulting in a three-tiered risk stratification: low (0–4 points), intermediate (5–7) and high (8–12), with respective median survivals of 56, 27.4 and 9.2 months.7 It should be noted that all nucleotide variations (missense, nonsense and frameshift) were regarded as ASXL1 mutations in the Mayo study,9 whereas only nonsense and frameshift ASXL1 mutations were considered in the French study.7
To further clarify the prognostic relevance of ASXL1 mutations, an international collaborative cohort of 466 CMML patients was analyzed.4 In univariate analysis, survival was adversely affected by ASXL1 (nonsense and frameshift) mutations. In multivariable analysis, ASXL1 mutations, AMC >10 × 109/l, HB<10 gm/dl, platelets <100 × 109/l and circulating IMC were independently predictive of shortened OS. A regression coefficient-based prognostic model (Molecular Mayo Model) based on these five risk factors delineated high (⩾3 risk factors; HR 6.2, 95% CI 3.7–10.4) intermediate-2 (2 risk factors; HR 3.4, 95% CI 2.0–5.6) intermediate-1 (1 risk factor; HR 1.9, 95% CI 1.1–3.3) and low (no risk factors) risk categories, with median survivals of 16, 31, 59 and 97 months, respectively.6
Efficient H3K27 methylation requires the corporation of core components, including EZH2 (catalytic enzyme) and cofactors SUZ12 and EED. The EZH2 (enhancer of zeste homolog 2—chromosome 7q35-q36) gene, encodes for the PRC2 protein, a highly conserved enzyme that serves as a histone H3K27 methyltransferase (Figure 5). In CMML, EZH2 mutations are infrequent (~5%) and do not have an independent prognostic impact.7, 59 The UTX gene (ubiquitously transcribed X chromosome tetratricopeptide repeat—chromosome Xp11.2), encodes a lysine-specific demethylase (6A). UTX mutations are seen in ~8% of CMML patients and do not impact survival.59
Spliceosome component mutations (SRSF2, SF3B1, U2AF1 and ZRSR2)
Spliceosome component mutations (SRSF2, SF3B, U2AF1 and ZRSR2) affect pre-mRNA splicing.16, 60 They are involved in the 3′ splice site recognition of pre-mRNA, including abnormal/alternative splicing. The U2 auxiliary factor that consists of the U2AF65–U2AF1 heterodimer establishes physical interaction with SF1 and a serine/arginine-rich protein such as SRSF1 or SRSF2, resulting in recognition of the 3′ splice site and its nearby polypyrimidine tract.60 This leads to the subsequent recruitment of U2 snRNP containing SF3A1 and SF3B1 to establish the splicing A complex.60
SRSF2 (serine/arginine-rich splicing factor 2—chromosome 17q25.2) mutations are seen in patients with MDS, CMML, PMF and AML.60, 61, 62, 63 In MDS and PMF, these mutations are seen in ~15–20% of patients and are associated with a shortened OS and LFS.61, 63, 64 In CMML, the frequency of SRSF2 mutations is higher (~50%), and these mutations are associated with increased age, less pronounced anemia and a diploid karyotype.16 Mutational hot spots include P95L, P95H and P95R.16 Thus far, in CMML, SRSF2 mutations have not demonstrated an independent prognostic impact on either OS or LFS.7, 16, 65
SF3B1 (splicing factor 3B, subunit 1—chromosome 2q33.1) mutations have a high prevalence (~80%) in patients with MDS and ring sideroblasts (RS)66 and can also be seen in patients with CMML and RS (<10%).16 In MDS and CMML, these mutations do not influence either the OS or LFS.63, 67 The mutational hot spots for SF3B1 include K700E (~50%), H662Q and K666N.16, 66 Gene expression studies have shown that SF3B1 mutations result in the downregulation of ABCB7 (ATP-binding cassette, sub-family B, member 7), a mitochondrial cassette protein, resulting in the development of RS.68
U2AF1 (U2 small nuclear RNA auxiliary factor—chromosome 21q22) mutations are seen in ~10% of patients with CMML and have thus far lacked an independent prognostic effect.60 The mutational hot spots for U2AF1 include S34F, Q157 and R158H.16 ZRSR2 mutations (zinc finger, RNA-binding motif and serine/arginine-rich factor—chromosome Xp22.2) are very infrequent and once again do not have an independent prognostic impact.60
Mutations involving the cohesin complex (STAG2, BCOR, SMC3, SMC1A and RAD21)
Cohesin is a multimeric protein complex composed of four core subunits: SMC1, SMC3, RAD21, and STAG proteins, together with a number of regulatory molecules, such as PDS5, NIPBL and ESCO.69 Cohesin is thought to be engaged in the cohesion of sister chromatids during cell division, postreplicative DNA repair and the regulation of global gene expression.70, 71 Germline mutations in cohesin components lead to the congenital multisystem malformation syndromes known as Cornelia de Lange syndrome and Roberts syndrome.70, 71
Mutations involving the cohesin complex can be seen in myeloid neoplasms, with individual mutational frequencies of ~12% AML, ~8% MDS, ~6% chronic myeloid leukemia, ~1% MPN and ~10% in CMML.69 These mutations frequently coexist with other myeloid relevant mutations, including TET2, ASXL1 and EZH2.69 The prognostic impact of these mutations remains to be determined.
Mutations involving DNA damage response genes (Tp53 and PHF6)
The PHF6 gene (plant homeodomain finger protein 6—chromosome Xp26.3) is a tumor-suppressor gene commonly mutated in T-cell acute lymphoblastic leukemia (~20%).72 PHF6 has two PHD domains involved in the recognition of epigenetic histone marks, which suggests a role in the epigenetic regulation of gene expression. PHF6 mutations are infrequent in chronic myeloid neoplasms, including CMML (~2.5%).
The Tp53 (tumor protein 53—chromosome 17p13.1) gene encodes a tumor-suppressor protein containing transcriptional activation, DNA binding and oligomerization domains. The encoded protein regulates the expression of target genes under stress, thereby inducing cell cycle arrest, apoptosis, senescence, DNA repair and changes in metabolism. Mutations in this gene are associated with a variety of human cancers, including hereditary cancer syndromes such as the Li-Fraumeni syndrome. Tp53 mutations are very infrequent in CMML (~1%).7
Mutations in transcription factors, signal transduction and cellular/receptor tyrosine kinase pathways (RUNX1, JAK2, KRAS, NRAS, CBL, SH2B3, FLT3)
The RUNX1 gene (runt-related transcription factor 1—chromosome 21q22.3) encodes the DNA-binding, alpha subunit of the core-binding factor and is essential for normal hematopoiesis and differentiation. It helps regulate the expression of G-CSF, interleukin-3, T-cell receptor and myeloperoxidase.73 Mutations and translocations involving RUNX1 have been well characterized in core-binding factor AML (t(8;21)(q22;q22) RUNX1/RUNX1T1) and MDS.73 In CMML, RUNX1 mutations are seen in ~15% of patients.7, 74, 75 These mutations do not impact OS but can be associated with a shorter LFS, especially in patients with C-terminal mutations.7, 74, 75
Signal pathway mutations are common in CMML and include: JAK2V617F (~10–15%), RAS (KRAS and NRAS ~20–30%), and CBL (~10–20%) mutations.7, 75 RAS (KRAS—-Kirsten Rat Sarcoma viral oncogene homolog—chromosome 12p12.1 and NRAS—Neuroblastoma RAS viral oncogene homolog—chromosome 1p13.2) mutations are often associated with a MPN-like CMML phenotype.76 Although univariate analysis studies with RAS mutations have demonstrated inferior outcomes, these findings have not been substantiated in multivariable models.7, 8
The CBL gene (casitas B-cell lymphoma—chromosome 11q23.3) codes for an E3 ubiquitin ligase involved in degradation of activated receptor tyrosine kinases, thereby resulting in a negative modulation of tyrosine kinase signaling.77 RING finger domain mutations of CBL are frequently associated with UPD11q (uniparental disomy) and have been reported in 10–20% of patients with CMML.7, 75, 77 CBL mutations are associated with monosomy 7 and TET2 mutations but, thus far, in CMML, have had no impact on OS or LFS.7, 77 SH2B3 (SH2B adaptor protein 3, also called as LNK—chromosome 12q24.12) is a key negative regulator of cytokine signaling and has a critical role in hematopoiesis. SH2B3 directly binds to wild-type JAK2 and JAK2 V617F and decreases their autophosphorylation and downstream signaling through STAT5 (signal transducer and activator of transcription factor 5), MAPK (mitogen-activated protein kinase)/ERK (extracellular signal–regulated kinase) and the PI3K (phosphoinositide-3 kinase)/AKT pathways.78 SH2B3 mutations are seen in ~5–7% of CMML patients and may co-occur with CBL mutations, suggesting a collaborative effect.79 These mutations, again, lack an independent prognostic effect on disease outcomes.
The FLT3 gene (Fms-like tyrosine kinase 3—chromosome 13q12.2) codes for a type III receptor tyrosine kinase that regulates differentiation, proliferation and survival of hematopoietic progenitor cells. The FLT3 ITD (internal tandem duplication) is found in ~30% of patients with cytogenetically normal AML and predicts poor outcomes.80 FLT3 mutations (ITD and tyrosine kinase domain mutations) are seen in <5% of patients with MDS and CMML and, unlike in AML, do not impact OS or LFS.7, 81 Mutations involving NPM1 (nucleophosphomin—chromosome 5q35.1) and c-Kit (chromosome 4q12) are very uncommon in CMML.7
SETBP1 mutations
SETBP1 (SET-binding protein 1—chromosome 18q21.1) encodes the SET-binding protein 1, a binding partner for the multi-function SET protein. This protein is involved in apoptosis, transcription and nucleosome assembly.82 The proposed functional outcome of this interaction is based on in vitro studies that demonstrate a protection of SET protein from protease cleavage that results in inhibition of protein phosphatase 2A activity, leading to higher rates of cell proliferation. In CMML, SETBP1 mutations have a frequency of 5–10%, with some82, 83 but not all studies demonstrating prognostic relevance.6
Molecular and cytogenetic correlates in CMML
In a seminal, international collaborative study (Mayo Clinic and the French CMML consortium), cytogenetic and molecular correlates were assessed in 409 patients with WHO-defined CMML.4 The mutational frequency of commonly affected genes was: SRSF2 (46%), ASXL1 (37%), U2AF1 (8%), SF3B1 (7%), and SETBP1 (4%), respectively. ASXL1 and SF3B1 mutations were associated with abnormal karyotypes (P=0.04 and P=0.03) and SRSF2 with normal karyotypes (P=0.02).4 In comparison to other abnormal karyotypes, the incidence of ASXL1 mutations was lower in -Y (P=0.04) and der(3q) (P=0.03). U2AF1 mutations were associated with monosomal karyotypes (P=0.03) and SF3B1 with der(3q) (P<0.0001).4 There were 9 (2%) patients with der(3q) abnormalities of which 6 had sole der(3q). Five of the 9 (55%) evaluable patients with der(3q) had SF3B1 mutations and expressed BM RS. Iron stains were not available in four patients. This study resulted in the development of the aforementioned Mayo–French cytogenetic risk stratification system.
Conclusions
CMML is a myeloid neoplasm with overlapping features of MDS and MPN, enriched with cytogenetic (~30%) and molecular abnormalities (>90%).2, 22 Common cytogenetic abnormalities include: trisomy 8, -Y, abnormalities of chromosome 7 (monosomy 7 and del7q), trisomy 21, del(20q) and complex karyotypes. The Mayo–French cytogenetic risk stratification system effectively risk stratifies CMML patients based on cytogenetic abnormalities.4 The advent of next-generation sequencing has identified multiple gene mutations in most CMML patients. These mutations tend to involve epigenetic regulator genes (TET2, ASXL1), splicing components (SRSF2), signal pathways (RAS, CBL) and transcription factors (RUNX1).6, 7 Among these, thus far, only nonsense and frameshift ASXL1 mutations have been shown to negatively impact OS.6, 7 Expanding molecular insights into pathways altered by the above-mentioned genetic and epigenetic changes are slowly but surely translating into pharmacological interventions. A prime example is the availability of IDH inhibitors for IDH1/2-mutated myeloid neoplasms.29 Hopefully with time, molecular testing at diagnosis will not only help with disease prognostication but will also help offer better therapeutic approaches. The need of the hour is to develop a CMML prognostic model that incorporates cytogenetic and molecular abnormalities.
References
Swederlow S, Camp E, Harris NL, Jaffe ES, Stefano PA, Stein H, et al. (eds). WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer: Lyon, France, 2008.
Patnaik MM, Parikh SA, Hanson CA, Tefferi A . Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol 2014; 165: 273–286.
Patnaik MM, Wassie EA, Lasho TL, Hanson CA, Ketterling R, Tefferi A . Blast transformation in chronic myelomonocytic leukemia: risk factors, genetic features, survival, and treatment outcome. Am J Hematol 2015; 90: 411–416.
Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. Am J Hematol 2014; 89: 1111–1115.
Such E, Cervera J, Costa D, Sole F, Vallespi T, Luno E et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica 2011; 96: 375–383.
Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia 2014; 28: 2206–2212.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 2013; 31: 2428–2436.
Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood 2002; 99: 840–849.
Patnaik MM, Padron E, LaBorde RR, Lasho TL, Finke CM, Hanson CA et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia 2013; 27: 1504–1510.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–3627, quiz 3699.
Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–2417.
Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med 2002; 347: 481–487.
Pardanani A, Ketterling RP, Li CY, Patnaik MM, Wolanskyj AP, Elliott MA et al. FIP1L1-PDGFRA in eosinophilic disorders: prevalence in routine clinical practice, long-term experience with imatinib therapy, and a critical review of the literature. Leuk Res 2006; 30: 965–970.
Curtis CE, Grand FH, Musto P, Clark A, Murphy J, Perla G et al. Two novel imatinib-responsive PDGFRA fusion genes in chronic eosinophilic leukaemia. Br J Haematol 2007; 138: 77–81.
Tefferi A, Gilliland DG . Oncogenes in myeloproliferative disorders. Cell Cycle 2007; 6: 550–566.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol 2013; 88: 201–206.
Tang G, Zhang L, Fu B, Hu J, Lu X, Hu S et al. Cytogenetic risk stratification of 417 patients with chronic myelomonocytic leukemia from a single institution. Am J Hematol 2014; 89: 813–818.
Patnaik MM, Hanson CA, Hodnefield J, Knudson R, Van Dyke D, Tefferi A . Monosomal karyotype in myelodysplastic syndromes, with or without monosomy 7 or 5, is prognostically worse than an otherwise complex karyotype. Leukemia 2010; 25: 266–270.
Patnaik MM, Lasho TL, Finke CM, Gangat N, Caramazza D, Holtan SG et al. WHO-defined 'myelodysplastic syndrome with isolated del(5q)' in 88 consecutive patients: survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia 2010; 24: 1283–1289.
Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D . Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 2012; 5: 12.
Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C et al. Clonal architecture of chronic myelomonocytic leukemias. Blood 2013; 121: 2186–2198.
McCullough KB, Patnaik MM . Chronic myelomonocytic leukemia: a genetic and clinical update. Curr Hematol Malig Rep 2015; 10: 292–302.
Itzykson R, Solary E . An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia 2013; 27: 1441–1450.
Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood 2013; 121: 5068–5077.
Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 2012; 7: 201–207.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360: 2289–2301.
Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009; 23: 1343–1345.
Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 2009; 23: 900–904.
Abdel-Wahab O, Levine RL . Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013; 121: 3563–3572.
Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009; 114: 144–147.
Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011; 473: 398–402.
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010; 468: 839–843.
Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010; 17: 13–27.
Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011; 20: 25–38.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011; 20: 11–24.
Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA 2011; 108: 14566–14571.
Patnaik MM, Lasho TL, Vijayvargiya P, Finke CM, Hanson CA, Ketterling RP et al. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J 2016; 6: e385.
Patnaik MM, Wassie EA, Padron E, Onida F, Itzykson R, Lasho TL et al. Chronic myelomonocytic leukemia in younger patients: molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J 2015; 5: e280.
Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014; 124: 2705–2712.
Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia 2012; 26: 101–105.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18: 553–567.
Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011; 25: 1153–1158.
Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011; 25: 1217–1219.
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 2011; 25: 1200–1202.
Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun 2015; 6: 7307.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012; 22: 180–193.
Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 2010; 24: 1062–1065.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 145: 788–800.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009; 23: 2183–2186.
Carbuccia N, Trouplin V, Gelsi-Boyer V, Murati A, Rocquain J, Adelaide J et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia 2010; 24: 469–473.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364: 2496–2506.
Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013; 27: 1861–1869.
Damaj G, Joris M, Chandesris O, Hanssens K, Soucie E, Canioni D et al. ASXL1 but not TET2 mutations adversely impact overall survival of patients suffering systemic mastocytosis with associated clonal hematologic non-mast-cell diseases. PLoS One 2014; 9: e85362.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–1089.
Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 2013; 27: 82–91.
Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood 2011; 118: 5593–5603.
Abdel-Wahab O, Kilpivaara O, Patel J, Busque L, Levine RL . The most commonly reported variant in ASXL1 (c.1934dupG;p.Gly646TrpfsX12) is not a somatic alteration. Leukemia 2010; 24: 1656–1657.
Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol 2011; 29: 2499–2506.
Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 2011; 118: 3932–3941.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478: 64–69.
Lasho TL, Jimma T, Finke CM, Patnaik M, Hanson CA, Ketterling RP et al. SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012; 120: 4168–4171.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011; 365: 1384–1395.
Aribi A, Borthakur G, Ravandi F, Shan J, Davisson J, Cortes J et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer 2007; 109: 713–717.
Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood 2012; 120: 3106–3111.
Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012; 120: 3080–3088.
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 2012; 119: 569–572.
Patnaik MM, Hanson CA, Sulai NH, Hodnefield JM, Knudson RA, Ketterling RP et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization–defined myelodysplastic syndromes without excess blasts. Blood 2012; 119: 5674–5677.
Gerstung M, Pellagatti A, Malcovati L, Giagounidis A, Porta MG, Jadersten M et al. Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat Commun 2015; 6: 5901.
Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013; 45: 1232–1237.
Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008; 451: 796–801.
Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008; 132: 422–433.
Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet 2010; 42: 338–342.
Speck NA, Gilliland DG . Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002; 2: 502–513.
Kuo MC, Liang DC, Huang CF, Shih YS, Wu JH, Lin TL et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia 2009; 23: 1426–1431.
Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 2010; 28: 3858–3865.
Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res 2010; 16: 2246–2256.
Schnittger S, Bacher U, Alpermann T, Reiter A, Ulke M, Dicker F et al. Use of CBL exon 8 and 9 mutations in diagnosis of myeloproliferative neoplasms and myelodysplastic/myeloproliferative disorders: an analysis of 636 cases. Haematologica 2012; 97: 1890–1894.
Bersenev A, Wu C, Balcerek J, Tong W . Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest 2008; 118: 2832–2844.
Koren-Michowitz M, Gery S, Tabayashi T, Lin D, Alvarez R, Nagler A et al. SH2B3 (LNK) mutations from myeloproliferative neoplasms patients have mild loss of function against wild type JAK2 and JAK2 V617F. Br J Haematol 2013; 161: 811–820.
Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008; 358: 1909–1918.
Daver N, Strati P, Jabbour E, Kadia T, Luthra R, Wang S et al. FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am J Hematol 2013; 88: 56–59.
Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet 2013; 45: 942–946.
Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia 2013; 27: 2100–2102.
Acknowledgements
Current study is supported in part by grants from the 'The Henry J. Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN, USA'. This publication was supported by CTSA Grant Number KL2 TR000136 from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We thank Dr Terra Lasho, PhD in helping prepare two of the figures for the manuscript (Figures 4 and 5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Patnaik, M., Tefferi, A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer Journal 6, e393 (2016). https://doi.org/10.1038/bcj.2016.5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2016.5
This article is cited by
-
Emergence of clone with PHF6 nonsense mutation in chronic myelomonocytic leukemia at relapse after allogeneic HCT
International Journal of Hematology (2022)
-
Mutational landscape of chronic myelomonocytic leukemia and its potential clinical significance
International Journal of Hematology (2022)
-
EZH2 inactivation in RAS-driven myeloid neoplasms hyperactivates RAS-signaling and increases MEK inhibitor sensitivity
Leukemia (2021)
-
Increasing recognition and emerging therapies argue for dedicated clinical trials in chronic myelomonocytic leukemia
Leukemia (2021)
-
Mutations in chronic myelomonocytic leukemia and their prognostic relevance
Clinical and Translational Oncology (2021)
