
Abstract
This prospective phase II study evaluated the efficacy of azacitidine (Aza)+erythropoietin (Epo) in transfusion-dependent patients with lower-risk myelodysplastic syndrome (MDS). Patients ineligible for or refractory to full-dose Epo+granulocyte colony stimulation factors for >8 weeks and a transfusion need of ⩾4 units over 8 weeks were included. Aza 75 mg m−2 d−1, 5/28 days, was given for six cycles; non-responding patients received another three cycles combined with Epo 60 000 units per week. Primary end point was transfusion independence (TI). All patients underwent targeted mutational screen for 42 candidate genes. Thirty enrolled patients received ⩾one cycle of Aza. Ten patients discontinued the study early, 7 due to adverse events including 2 deaths. Thirty-eight serious adverse events were reported, the most common being infection. Five patients achieved TI after six cycles and one after Aza+Epo, giving a total response rate of 20%. Mutational screening revealed a high frequency of recurrent mutations. Although no single mutation predicted for response, SF3A1 (n=3) and DNMT3A (n=4) were only observed in non-responders. We conclude that Aza can induce TI in severely anemic MDS patients, but efficacy is limited, toxicity substantial and most responses of short duration. This treatment cannot be generally recommended in lower-risk MDS. Mutational screening revealed a high frequency of mutations.
Similar content being viewed by others

Introduction
The myelodysplastic syndromes (MDSs) constitute a group of malignant hematopoietic stem cell disorders characterized by pancytopenia, ineffective hematopoiesis, dysplastic bone marrow changes and a significant risk for progression to acute myeloid leukemia (AML).
Anemia is the dominating symptom in low- and intermediate-1-risk MDS. Around 50% of these patients respond to growth factor treatment, that is, erythropoiesis-stimulating agents (ESA)±granulocyte colony stimulation factors (G-CSF), and the median duration of response is 2 years.1, 2, 3, 4 A predictive model based on serum levels of erythropoietin (S-Epo) and the red blood cell transfusion rate was developed to select patients with different probabilities for response to treatment. Patients with a transfusion need of ⩾2 red blood cell units per 4 weeks and S-Epo level >500 U l−1 showed only 7% response and were considered not eligible for this treatment.5 Two large retrospective studies indicate an improved survival for patients treated with ESA compared with untreated patients.2, 6 Patients not eligible for or refractory to growth factors require transfusion therapy with compromised quality of life possibly need for iron chelation therapy and worse outcome in case of allogeneic stem cell transplantation.7
In Europe, azacitidine (Aza) is registered for treatment of patients with higher-risk MDS where it has been shown to prolong survival.8 In the US, the label encompasses all patients with MDS. A subgroup analysis of patients (n=22) with refractory anemia/refractory anemia with ringed sideroblast in the pivotal, prospective study showed complete remission, partial remission and hematological improvement in 9, 18 and 32%, respectively.9 A second prospective study reported transfusion independence (TI) after treatment with Aza in 65% of previously transfusion-dependent (TD) patients (n=40) with lower-risk MDS.10 Furthermore, two retrospective studies evaluated the effect of Aza in lower-risk MDS populations. Musto et al.11 reported complete remission, partial remission and hematological improvement was 16, 10 and 20%, respectively, and Prebet et al.12 reported complete remission, partial remission and hematological improvement was 12, 5 and 25%, respectively. None of these four studies employed ESA resistance as an inclusion criterion. Recently, a prospective study treating ESA-resistant patients with lower-risk MDS with Aza 75 mg m−2 per day 1–5 every 28 days showed overall response rate of 47% and achievement of TI in previously TD patients in 33% of the patients.13
Mutations in a number of genes have been linked to the MDS disease, and some of them also with outcome.14 A recent study of 288 patients with low- and Int-1-risk MDS identified TET2 (23% of samples), SF3B1 (22%), U2AF1 (16%), ASXL1 (15%), SRSF2 (15%) and DNMT3A (13%) as the most commonly mutated genes in a cohort of lower-risk MDS.15 Early association studies between mutational status and response to Aza indicate a higher response rate for TET2-mutated patients and a lower response rate for ASXL1-mutated patients.16, 17
We hypothesized that Aza can induce TI in TD, ESA-resistant patients with lower-risk MDS and enrolled 30 patients in an investigator-initiated prospective phase II trial. Aza was assessed as monotherapy and in combination with ESA. Compared with previous reports, the overall efficacy of Aza in our study was limited and toxicity considerable. Targeted sequencing revealed a high frequency of recurrent gene mutations, most commonly involving splice factor genes and epigenetic regulators.
Subjects and methods
Study population
Eligible patients should fulfill the inclusion criteria: (i) age of 18 years or more; (ii) MDS with international prognostic score system (IPSS) low or Int-1, or mixed MDS/myeloproliferative disorder; either CMML with <10% marrow blasts or refractory anemia with ringed sideroblast-T; (iii) patients with high or intermediate probability for response according to the predictive model5 should be refractory to Epo (equivalent to ⩾60 000 U of EPO per week for ⩾8 weeks), followed by Epo+G-CSF for ⩾8 weeks, biosimilar drugs in equipotent doses or Epo+G-CSF upfront for 8 weeks. Patients with low probability for response according to the predictive model5 were eligible without prior Epo/G-CSF treatment. Assessment of refractoriness was performed at the time of enrollement; (iv) transfusion rate ⩾4 units over the last 8 weeks or ⩾8 units over the last 26 weeks; (v) subject had signed the informed consent document; and (vi) men and women of childbearing potential had to use effective contraception during and three months after treatment. Exclusion criteria encompassed eligibility for curative treatment and are listed in Supplementary Table 1.
The trial was approved by the National Ethical committees of Sweden, Denmark and Norway, conducted in accordance with the Declaration of Helsinki and registered at www.clinicaltrials.gov: NCT01048034. The Nordic MDS Group was the sponsor of this trial, for which Celgene provided a research grant and the study drug Aza, and Roche provided the study drug beta-Epo.
Study protocol is present in Supplementary Data.
Study design
The main objective of this prospective open-label, non-randomized, multicenter phase II trial was to study the efficacy of Aza to induce TI in previously TD patients. For this reason, number of patients achieving TI was chosen as the primary end point. Secondary objectives encompassed TI on the combination of Aza+ESA, effect on bone marrow morphology and biology, peripheral blood parameters, cytogenetics, safety and response in relation to mutational status. Enrolled patients received six cycles of Aza, 75 mg m−2 for 5 consecutive days every 28 days. Patients achieving TI terminated the study after six cycles and were subject to maintenance treatment according to the investigator’s judgement. The schedule and dosage of the maintenance treatment was designed by the investigator. Patients still TD after 6 cycles continued with another three cycles of Aza, with the addition of Epo-β 60 000 units per week. Efficacy assessment was performed after six cycles (week 28) and nine cycles (week 45). Toxicity, including cytopenias, was graded according to the National Cancer Institute toxicity criteria (CTC for Adverse Events, version 3.0, Washington DC, WA, USA). In case of suspected drug-related hematological toxicity, the dose of Aza could be lowered according to a pre-defined scheme. In case of severe hematological toxicity (absolute neutrophil count (ANC)<0.5 × 109 l−1 or platelet (Plt)<15 × 109l−1) or non-hematological toxicity grade 3 or 4, the next planning cycle was temporarily discontinued.
Use of G-CSF was allowed during treatment when found appropriate by the investigator.
Morphology and cytogenetics
Bone marrow morphology, including biopsy for assessment of cellularity and fibrosis, was performed before inclusion and at week 28 and 45. Morphological specimens were reviewed centrally according to the standard procedure of the Nordic MDS group.18 The morphological assessment included presence of dysplasia, blast count, cellularity and fibrosis. Metaphase cytogenetic analysis performed within 6 months before inclusion was used to establish the karyotype. Assessment of cytogenetic response was performed at week 28 if chromosomal aberrations were present at inclusion.
Mutational analysis
All the patients registered for this study were analyzed for 42 genes recurrently mutated in myeloid disorders using high throughput sequencing by Haloplex target enrichment technology (Agilent Technologies, Sanata Clara, CA, USA), see Supplementary Table 5 for a list of included genes. In general, CD34+ cells were separated from bone marrow aspirates pre-treatment by a magnetic activated cell sorting (MACS, Miltenyi Biotec Norden, Lund, Sweden) as previously described.18 Genomic DNA was extracted from 1 × 106 CD34− cells using the Gene Elute genomic DNA extraction kit (Sigma-Aldrich, Stockholm, Sweden). The Haloplex target enrichment kit G9901A/B was designed using Halodesign wizard available by Agilent (http://www.halogenomics.com), 679 target regions were included with 99.2% coverage of the 42 selected genes. All samples were individually barcoded during enrichment and samples were pooled before sequencing using Illumina HiSeQ 2000 system (Illumina, San Diego, CA, USA). Sequencing reads were mapped over human genome 19 and the variants were called using SAMTOOLS.19 The minimum coverage to consider in the analysis was 30 and the allele should have a minimum of 10% of reads. Sequence variations were annotated and functionally classified using ANNOVAR.20 Variants previously reported as germline polymorphisms in 1000 genome and ESP5400 databases were excluded.21, 22 Finally, variants located in non-coding regions as well as synonymous variants were filtered out.
Response criteria
TI was defined as no need for red blood cell transfusions in combination with a stable hemoglobin during the 8 weeks preceding evaluation. Erythroid response was also assessed using the International Working Group criteria for MDS,23 for example, an absolute reduction of the transfusion need by ⩾4 units per 8 weeks. In addition, morphological response, platelet response and neutrophil response were assessed using the International Working Group criteria.23 Complete cytogenetic response was defined as normalized karyotype by metaphase cytogenetic analysis for patients with previous aberrations. Safety was evaluated by monitoring and recording of adverse events.
Study evaluation and statistics
The study was designed to detect an efficacy of ⩾20%, defined as ⩾20% of patients reaching the primary end point. By using Wald's sequential probability ratios, we defined the number of treated patients needed to conclude, with a power of 90% and an alpha error of 5%, that the treatment was inefficacious, for example, has an efficacy of <20%. If 14 consecutive patients were to fail to respond to treatment, the study was planned to close for further inclusion.
The response population for Aza as monotherapy was defined as all patients receiving ⩾1 dose of Aza. In addition, response rate was also calculated for the patients completing six cycles of Aza. Similarly, the response population for the combined treatment of Aza+ESA consisted of all patients receiving one additional dose of Aza combined with ESA, and response rates were also calculated for patients completing three cycles of the combined treatment. The safety population consisted of all included patients.
Continuous variables were summarized using the mean or median (range) value depending on distribution of data and frequency tables were used to summarize categorical variables.
Association between pre-treatment characteristics and response was assessed using a univariate approach. Statistical methods used were t-test for normally distributed data, Mann–Whitney U-test for non-normally distributed data and Fisher’s exact test for categorical data. The sample size did not allow for multivariate testing.
All statistical calculations were performed using the SPSS 20.0 software (IBM, New York, NY, USA).
Long-term follow-up
Enrolled patients are evaluated regarding survival, transformation to AML and subsequent treatment for up to 3 years after inclusion.
Results
Patient characteristics
Thirty patients from 11 centers were enrolled between January 2010 and September 2011 including 21 men and 9 women with a median age of 69 years. The most common WHO subclasses were refractory cytopenia with multilineage dysplasia (n=13) and refractory cytopenia with multilineage dysplasia-RS (n=5) (see Table 1 for details). An additional six patients were screen failures. Median duration of disease was 2 years (range 0–20). Median number of transfused units of erythrocytes during the 8 and 26 weeks preceding inclusion was 7 (range 4–14) and 16 (range 4–40), respectively. Median bone marrow cellularity and blast count was 70% (20–100) and 2% (0–9), respectively. Six patients scored IPSS low, whereas 24 patients scored intermediate-1. Twenty patients belonged to the favorable IPSS cytogenetic risk group, whereas 10 patients belonged to the intermediate group. Eighteen patients were previously treated with ESA+G-CSF without achieving TI, remaining patients had poor probability of response according to the predictive model and were considered refractory upfront. Median Plt and ANC pre-treatment were 210 × 109 l−1 (range 22–1468) and 1.7 × 109 l−1 (range 0.3–15.1), respectively. Thrombocytopenia (Plt count <100 × 109l−1) was observed in five patients, before start of treatment, of which three patients had Plt counts <30 × 109l−1. Neutropenia (<1.0 × 109l−1) was observed in eight patients before start of treatment including four cases with <0.5 × 109l−1. See Supplementary Table 4 for characteristics of each patient.
Frequency of mutations
Sequencing was performed in 27 patients of whom 24 had one or more mutations (Figure 1). Of the three patients without mutations, two had a chromosomal aberration. Hence, 96% of the patients had at least one clonal marker. A total number of 53 mutations were identified and the median number of mutations per patient was 2 (range 0–5). Nineteen and 17 patients had a mutation involving a splice factor gene or an epigenetic regulator, respectively. Two and four patients had mutations in more than one splice factor gene and epigenetic regulator, respectively. The most frequently mutated genes were SF3B1 (n=11), ASXL1 (n=9) and TET2 (n=7). All patients with refractory anemia with ringed sideroblast or refractory cytopenia with multilineage dysplasia-RS (n=7) had a mutated SF3B1; three and two patients had a concomitant TET2 and DNMT3A mutation, respectively. The number of mutations did not correlate with disease duration (P=0.157). See figure 1 and Supplementary Table 2 for detailed information.

Illustration of mutational and cytogenetic status for included patients.
Treatment results
Thirty patients received ⩾1 cycle of Aza and 22 of these completed the initial six cycles of Aza as monotherapy. Eight patients pre-terminated the treatment after cycle 1 (n=1), cycle 2 (n=2), cycle 3 (n=3) and cycle 5 (n=2). Sixteen patients continued with the combined treatment of Aza+ESA, 15 of which completed the three additional cycles. Of 30 patients receiving ⩾1 cycle of Aza as monotherapy, 5 (17%) achieved TI. Of the 16 patients receiving ⩾1 cycle of the combined treatment, 1 additive patient (6%) achieved TI. Response rates for patients completing all cycles of monotherapy and combined treatment were 5/22 (23%) and 1/15 (7%), respectively. An illustration of outcome for included patients is given in Figure 2a.

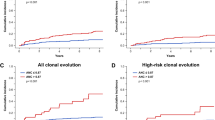
(a) Outcome after azacitidine as monotherapy and the combined treatment of azacitidine and erythropoietin (Epo). Response is defined as transfusion independence. (b) Duration of response. (c) Overall survival. (d) Leukemic transformation.
Three patients achieved an erythroid improvement (reduced transfusion requirement) in accordance to the International Working Group criteria, two on monotherapy and one on combined treatment. Hence, the total erythroid response rate was 9 of 30 (30%).
Four patients had an elevated blast count before treatment; three of these were evaluated after treatment, in which all three patients normalized their blast count. Only one of these patients achieved TI. Complete cytogenetic response was observed in one of seven evaluated patients, a patient with trisomy 8 who also achieved TI. Platelet response was not observed in any of the five patients with pre-treatment platelet count <100 × 109l−1. Neutrophil response was observed in one of eight patients with pre-treatment neutrophil count <1.0 × 109l−1. This patient also achieved TI.
Duration of response for the six responding patients was <6 months for four patients; one patient responded for 24 months and one patient is still responding after 21 months. Both long-term responders and one of the short-term responders were exposed to Aza given as maintenance treatment. Median number of cycles until TI was 3½. Duration of response for the three patients with hematologic improvement without complete TI was 2, 3 and 8 months, respectively. See Table 2 for details regarding responding patients.
Markers for response to treatment
Disease duration was significantly associated with response; responding patients had a shorter median disease duration compared with non-responders (0.5 vs 2 years, P=0.013). Unexpectedly, the six responders showed a higher median transfusion rate before treatment compared with non-responders (8.5 (range 4–10) vs 6 units (range 4–14), P=0.038). Moreover, all six patients had IPSS Int-1-risk MDS, whereas no IPSS low-risk patients responded. No other parameters such as morphology, karyotype, blood test including iron status and mutational status were associated with a response. Two of six responding patients had trisomy 8 compared with one of 24 non-responding patients.
TET2 was mutated in two (33%) of the responding (n=6) patients compared with five (28%) of the non-responding (n=21) patients (P=0.633). ASXL1 was mutated in three (50%) and six (29%) of the responding and non-responding patients, respectively (P=0.367). Although none of the mutations were significantly associated with response to treatment, certain mutations including DNMT3A (n=4) and SF3A1 (n=3) were only observed in non-responders. See Table 3 for a list of all tested variables.
Long-term outcome
Twelve patients have died during or after the study after a median follow-up of 30 months. Median survival is still not reached. Three patients (10%) transformed to AML during the follow-up period at 11, 13 and 27 months. See figure 2 for Kaplan–Meier estimation of survival and leukemic progression. Hence, survival and transformation rates were similar to what would be expected from an Int-1-risk cohort.
Safety evaluation
Ten patients pre-terminated the study: 5 due to sustained neutropenia or thrombocytopenia, 2 due to death, 2 due to patient’s wish and 1 due to investigators’ choice. No patient progressed to AML during the study. Dose reduction of Aza was needed in 11 patients. Ten patients had a temporary discontinuation of Aza of more than 1 week’s duration.
The most important hematological toxicity was neutropenia. Nineteen patients suffered from severe neutropenia (ANC⩽0.5 × 109l−1) at any time point during treatment, 4 of which were severely neutropenic before the treatment was started. Similarly, 11 patients suffered from severe thrombocytopenia (Plt⩽30 × 109l−1) during treatment, of which 3 were severely thrombocytopenic before start of treatment. No severe bleeding was reported in the study. Nadir values after a cycle of Aza were seen at week 3 for thrombocytes (median 115 × 109l−1) and at week 4 for neutrophils (median 1.1 × 109 l−1), respectively.
The most commonly reported non-hematological adverse events were infection (n=30) and the related adverse events were neutropenic fever (n=12) and fever (n=6). Other frequent non-hematological adverse events were injection-site reaction (n=14) and constipation (n=15). Two patients experienced atrial fibrillation and one ventricular tachycardia. No serious unexpected toxicity was observed.
Thirty-eight serious adverse events were reported in 18 patients during the study period. The main serious adverse event criterion (n=36) was in-patient hospitalization. The vast majority (n=28) of the serious adverse event were related to infection with or without neutropenia. A complete list of all reported hematological and non-hematological adverse events is enclosed in Table 4 and Supplementary Table 3.
Cause of death for the first patient is unknown; the patient suffered a sudden death after two cycles of Aza and had a moderate cytopenia before start of treatment, which was aggravated during treatment. Cause of death for the second patient was septicemia with Escherichia coli during Aza-associated severe neutropenia.
Ten patients received G-CSF due to neutropenia during the study with a median ANC level of 0.3 × 109l−1 (range 0.1–1–5) when G-CSF was started.
Discussion
This prospective study evaluated the efficacy of Aza in a population of selected ESA-resistant, TD patients with lower-risk MDS. It is an important study as Aza is used in clinical practice for lower-risk patients, as demonstrated recently by data from the French compassionate program. However, previous clinical trials of Aza in lower-risk patients have not specifically targeted Epo-/G-CSF-resistant cohorts. Patients enrolled in the present trial had a severe transfusion need, reflected in a high median transfusion demand before inclusion and a high median ferritin level. This study shows a lower response rate in terms of TI compared with previous reports. Moreover, only 2 of 30 patients showed responses lasting more than 6 months. The recent report by Fili et al.13 assessed Aza in a similar cohort of patients, however, that cohort included also non-TD patients, patients with a lower transfusion rate and enrolled patients who did not have the same degree of documented resistance against the combined EPO/G-CSF treatment. Fili et al. found an overall response rate of 47%, although only 27% of previously TD patients achieved TI. The response rate in our study is lower than that of previous reports, possibly due to the careful selection of documented ESA-/G-CSF-resistant patients.
Toxicity and most importantly neutropenia with associated infections were substantial in this study. The marked suppression of ANC and Plt counts observed during treatment cycles likely reflects a cytotoxic effect on the hematopoiesis. Most studies on lower-risk MDS have used the same Aza dose, 75 mg m−2 × V, which makes suboptimal dosing a less likely.
Interestingly, long-term follow-up demonstrated survival times and AML transformation rates as expected according to the risk profile (IPSS-R and WPSS, WHO-based prognostic score system) of the cohort. After a median follow-up of 30 months, median survival was reached neither for all included patients nor for non-responding patients. This is in contrast to previous reports on survival after Aza failure in lower-risk MDS: 15 months (n=59) and 17 months (n=423).12, 24 Moreover, only three patients transformed to AML during follow-up, which makes non-leukemic death the major cause of death for these patients. From a biological point of view, our cohort is thus characterized by a highly ineffective erythropoiesis but with a relatively low propensity of transformation to more advanced MDS.
No individual morphological, cytogenetic or genetic parameter was significantly associated with response to treatment. Larger studies will be needed to further assess potential biomarkers. Interestingly, two out of three patients with trisomy 8 responded; a favorable response rate for trisomy 8 patients has previously been reported in higher-risk MDS.25
Recurrent mutations were found in all but three sequenced patients, rendering this lower-risk cohort a mutational frequency at least as high as in previous reports on MDS of all risk groups.14, 15, 26, 27 A majority of the patients carried a mutation in a splice factor gene and/or in one or several of the genes involved in epigenetic regulation. None of the mutations predicted for a response to treatment. Notably, mutations in two genes DNMT3A (n=4) and SF3A1 (n=3) were only observed in non-responders, which deserve further investigation. The genomes of AML patients with DNMT3A mutations have previously been reported to be hypomethylated, which might explain why treatment with Aza is ineffective.28 These data differ to some extent from the preliminary reported data from Bejar et al.,17 showing a higher response rate for TET2-mutated patients and a lower response rate for ASXL1-mutated patients with low- and high-risk MDS. This discrepancy supports the need for much larger datasets to reach conclusive interpretations. In addition, the sharper response criteria, TI, used in this study may have contributed to the observed pattern.
We conclude that Aza can induce TI in a minor proportion of ESA-refractory lower-risk MDS patients, but that the efficacy overall is low with few long-lasting responses. Moreover, toxicity is significant with potentially deleterious outcome. Hence, Aza cannot be recommended as standard treatment for these patients and does not seem to synergize with ESA. It is possible that molecular analysis to some extent may assist in the decision making for this patient cohort. However, larger studies are needed to evaluate the impact of mutational status on response rate.
References
Jadersten M, Montgomery SM, Dybedal I, Porwit-MacDonald A, Hellstrom-Lindberg E . Long-term outcome of treatment of anemia in MDS with erythropoietin and G-CSF. Blood 2005; 106: 803–811.
Park S, Grabar S, Kelaidi C, Beyne-Rauzy O, Picard F, Bardet V et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood, United States 2008; 111: 574–582.
Greenberg PL, Sun Z, Miller KB, Bennett JM, Tallman MS, Dewald G et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood 2009; 114: 2393–2400.
Kelaidi C, Park S, Sapena R, Beyne-Rauzy O, Coiteux V, Vey N et al. Long-term outcome of anemic lower-risk myelodysplastic syndromes without 5q deletion refractory to or relapsing after erythropoiesis-stimulating agents. Leukemia 2013; 27: 1283–1290.
Hellstrom-Lindberg E, Negrin R, Stein R, Krantz S, Lindberg G, Vardiman J et al. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: proposal for a predictive model. Br J Haematol 1997; 99: 344–351.
Jadersten M, Malcovati L, Dybedal I, Della Porta MG, Invernizzi R, Montgomery SM et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol 2008; 26: 3607–3613.
Armand P, Kim HT, Cutler CS, Ho VT, Koreth J, Alyea EP et al. Prognostic impact of elevated pretransplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood 2007; 109: 4586–4588.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10: 223–232.
Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002; 20: 2429–2440.
Lyons RM, Cosgriff TM, Modi SS, Gersh RH, Hainsworth JD, Cohn AL et al. Hematologic response to three alternative dosing schedules of azacitidine in patients with myelodysplastic syndromes. J Clin Oncol 2009; 27: 1850–1856.
Musto P, Maurillo L, Spagnoli A, Gozzini A, Rivellini F, Lunghi M et al. Azacitidine for the treatment of lower risk myelodysplastic syndromes: a retrospective study of 74 patients enrolled in an Italian named patient program. Cancer 2010; 116: 1485–1494.
Prébet T, Thepot S, Gore SD, Dreyfus F, Fenaux P, Vey N . Outcome of patients with low-risk myelodysplasia after azacitidine treatment failure. Haematologica 2013; 98: e18–e19.
Fili C, Malagola M, Follo MY, Finelli C, Iacobucci I, Martinelli G et al. Prospective phase II study on 5-days azacitidine (5d-AZA) for treatment of symptomatic and/or erythropoietin unresponsive patients with low/int-1 risk myelodysplastic patients. Clin Cancer Res 2013; 19: 3297–3308.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364: 2496–2506.
Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 2012; 30: 3376–3382.
Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011; 25: 1147–1152.
Bejar RSK, Stojanov P, Zaneveld JE, Bar-Natan M, Caughey B . Next-generation sequencing of 213 MDS patient samples identifies mutation profiles associated with response to hypomethylating agents and overall survival. Leuk Res 2013; 37: S19–S20, (abstract O-024).
Möllgård L, Saft L, Treppendahl MB, Dybedal I, Nørgaard JM, Astermark J et al. Clinical effect of increasing doses of lenalidomide in high-risk myelodysplastic syndrome and acute myeloid leukemia with chromosome 5 abnormalities. Haematologica 2011, 2011-07-01 96: 963–971.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA et al. A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science, [Research Support, N.I.H., Extramural] 2012; 337: 64–69.
Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–425.
Garcia-Manero G, Xiao L, Najla AA, Mishra A, Padron E, Lancet JE et al. Outcome of patients (pts) with low and intermediate-1 risk myelodysplastic syndrome (MDS) after hypomethylating agent (HMA) failure. Blood 2013; 122: 388.
Grovdal M, Khan R, Aggerholm A, Antunovic P, Astermark J, Bernell P et al. Negative effect of DNA hypermethylation on the outcome of intensive chemotherapy in older patients with high-risk myelodysplastic syndromes and acute myeloid leukemia following myelodysplastic syndrome. Clin Cancer Res 2007; 13: 7107–7112.
Mian SA, Smith AE, Kulasekararaj AG, Kizilors A, Mohamedali AM, Lea NC et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica 2013; 98: 1058–1066.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–3627.
Qu Y, Lennartsson A, Gaidzik VI, Deneberg S, Bengtzen S, Arzenani MK et alGenome-Wide DNA Methylation Analysis Shows Enrichment of Differential Methylation in ‘Open Seas’ and Enhancers and Reveals Hypomethylation in DNMT3A Mutated Cytogenetically Normal AML (CN-AML), 16/11/2012. ASH meeting abstract, American Society of Hematology, 2012.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author Contributions
MT, LM and EH-L collected and analyzed data and wrote the manuscript. MT, ID, MG, MSH, LM and EH-L designed the study. LB, ID, HG, MG, EHL, IHD, KG, CM, LM, LN, AOK, AP, LS, MSH and MT provided clinical care to patients, assisted in the analysis of data and coauthored the paper. The Nordic MDS Group was the sponsor of this trial, for which Celgene and Roche provided the study drugs and Celgene provided a research grant.
Supplementary Information accompanies this paper on Blood Cancer Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tobiasson, M., Dybedahl, I., Holm, M. et al. Limited clinical efficacy of azacitidine in transfusion-dependent, growth factor-resistant, low- and Int-1-risk MDS: Results from the nordic NMDSG08A phase II trial. Blood Cancer Journal 4, e189 (2014). https://doi.org/10.1038/bcj.2014.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2014.8
This article is cited by
-
Current Therapeutic Landscape in Lower Risk Myelodysplastic Syndromes
Current Treatment Options in Oncology (2023)
-
New Approaches to Myelodysplastic Syndrome Treatment
Current Treatment Options in Oncology (2022)
-
Myeloid leukemoid reaction after initial azacitidine therapy for chronic myelomonocytic leukemia
International Journal of Hematology (2022)
-
More is less, less is more, or does it really matter? The curious case of impact of azacitidine administration schedules on outcomes in patients with myelodysplastic syndromes
BMC Hematology (2018)
-
Systematic review of azacitidine regimens in myelodysplastic syndrome and acute myeloid leukemia
BMC Hematology (2018)
