
Abstract
Multiple myeloma (MM) is a clonal plasma cell malignancy that is initiated by a number of mutations and the process of disease progression is characterized by further acquisition of mutations. The identification and functional characterization of these myelomagenic mutations is necessary to better understand the underlying pathogenic mechanisms in this disease. Recent advancements in next-generation sequencing have made the identification of most of these mutations a reality. However, the functional characterization of these mutations has been hampered by the lack of proper and efficient tools to dissect these mutations. Here we explored the possible utility of transcription activator-like effector nuclease (TALEN) genome engineering technology to tailoring the genome of MM cells. To test this possibility, we targeted the HPRT1 gene and found that TALENs are a very robust and efficient genome-editing tool in MM cells. Using cotransfected green fluorescent protein as an enrichment marker, single-cell subclones with desirable TALEN modifications in the HPRT1 gene were obtained in as little as 3–4 weeks of time. We believe that TALENs will greatly facilitate the functional study of somatic mutations in MM as well as other cancers.
Similar content being viewed by others


Introduction
Cancer arises from a single normal cell that undergoes further progression through sequential acquisition of various pathogenic mutations (depicted in Figure 1a). As next-generation sequencing technology becomes more affordable, the number of mutations identified in various cancers continues to increase. In fact, the most frequent mutations in all major cancers have now been mapped and cataloged.1, 2 Some of these mutated genes, for example, TP53, KRAS, BRAF, are well-known tumor suppressor genes or oncogenes, whereas the significance of mutations in other genes remains largely unknown. Although cancer is categorically clonal, tremendous subclonal heterogeneity is the unifying characteristic of all cancers due to prevalent mutation-driven clonal evolution.3, 4 In this post-genome sequencing era, the biggest question we are facing is how these mutations individually and in combination drive the initiation and progression of cancer.

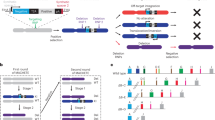
Cartoon depiction of the genesis of tumors. (a) Generation of tumors such as MM driven by sequential acquisition of various pathogenic mutations in patients. (b) Reconstruction of cancer mutations in mice by gene targeting approach is laborious and often nonproductive due to adverse phenotypes or not feasible due to the limited number of gene mutations that can be imposed (represented by the dashed line). (c) Our proposed TALEN-mediated deconstruction of cancer gene mutations in cancer cells such as HMCLs. Due to the method’s flexibility, the TALEN approach can create or remove mutations as indicated by the multiple arrows.
Theoretically, there are two ways to study cancer mutations in vivo. The first approach is to reconstruct cancer mutations in mice through homology-directed gene targeting, often referred to as gene knockouts or gene knockins.5 This approach replaces the endogenous locus of interest with a foreign DNA fragment that contains sequence homology on each end. This targeting approach has been successfully used in the past 20 years in mouse embryonic stem cells to generate mouse models. The major limitation of this gene targeting approach in mice is that only a limited number of mutated genes can be constructed in the mouse models through labor-intensive, time-consuming and expensive breeding strategies that intercross specific genetically manipulated mouse strains. Most importantly, the majority of mouse models do not faithfully phenocopy human diseases (depicted in Figure 1b).
The second way to study mutations in cancer is to deconstruct cancer mutations housed in human cancer cells from patients in reverse order by restoring the normality of these genes or by re-creating the mutations (Figure 1c). However, the very same gene targeting approach used in somatic cells (or somatic knockout) is also notoriously inefficient and time consuming.6, 7 There are only a handful somatic knockout cell lines in existence to this day. To that end, we believe a more efficient and flexible genome-editing method is urgently needed to revitalize the use of naturally occurring human cancer models, that is, human cancer cell lines, for functional studies of specific cancer mutations.
Recently, transcription activator-like effector nucleases (TALENs) have emerged as a highly effective tool for genome editing in various systems.8, 9, 10, 11 A pair of designed TALENs specifically recognize nearby DNA sequences in the gene of interest on opposite strands. The targeting results in the dimerization of the FokI nuclease cleavage domain in the TALEN modules, which then cleave the DNA sequence between the targeting sites, leading to double-stranded DNA breaks in the target gene. The double-stranded DNA break lesion is then repaired primarily by the error-prone non-homologous end-joining DNA repair system. Typically, after TALEN editing and DNA repair, the target gene is efficiently disrupted through shifting of the reading frame. In addition to gene disruption, the same approach can be used to remove or modify pre-existing mutations by providing exogenous DNA with mutation-corrected DNA sequences. Because of its flexible modular design, TALEN technology can precisely target almost any gene of interest, making this approach highly suitable for editing cancer cell lines, thereby enabling functional analysis of complex cancer mutations.
To prove the principle that TALEN technology can be used to tailor the genome of human cancer cell lines, we used multiple myeloma (MM) as a representative cancer. MM is an incurable and fatal malignancy that originates from post-germinal center plasma cells. MM is also an excellent cancer model to study because it is preceded by an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance and the development of MM involves sequential acquisition of various gene mutations (Figure 1a). Of note, the precise roles that various mutations acquired during the transition from benign to malignant disease remain unknown in many cases. Thus, if TALENs can be used in MM cells, this technology would be a powerful tool to study mutated gene function in this disease. The goal of this study, therefore, was to determine whether TALEN technology could be used to modify the genome of human myeloma cell lines (HMCLs). We chose to first target the HPRT1 gene and show here that TALENs are indeed robust genome-editing tools for HMCLs.
Materials and methods
TALEN design and construction
The 15-15-15 TALEN design strategy was employed to generate TALENs for this project.12 Using Mojohand (http://talendesign.org),10 two 15-mer repeat variable di-residue (RVD) TALENs with a 15-bp spacer were designed to target exon 3 of the human HPRT1 locus (Figure 2a). The 15-bp spacer contained an XhoI site that could be used to screen for TALEN cutting efficacy by restriction fragment length polymorphism analysis. A frameshift mutation at this location is expected to disrupt the phosphoribosyltransferase-type I domain that is essential to HPRT1 function.

TALEN structure and timeline for TALEN-mediated modification. (a) Sequence representation of TALEN-mediated HPRT1 gene targeting. PF and PR are PCR primers. (b) Timeline of the entire procedure shows its unmatched efficiency.
All TALEN constructs were synthesized with the Golden Gate method using the pC-GoldyTALEN scaffold (Addgene, Cambridge, MA, USA) described in Carlson et al.13 The repeat RVDs NI, HD, NG and NN (recognizing A, C, T and G bases, respectively) were used to construct TALENs targeting the HPRT1 locus. In the first Golden Gate reaction, RVDs 1 through 10 were synthesized in pFUS_A. Pre-synthesized pFUS_B4 plasmids were selected from an in-house library of all the possible combinations of 4-RVD pFUS_B plasmids (256 in total).12 The completed pFUS_A and pFUS_B4 as well as the last half-repeat plasmid (pLR-NI, -HD, -NN or -NG) were combined in the second Golden Gate reaction in the pC-GoldyTALEN expression vector that has a mini Caggs promoter.12
The pFUS_B4 collection has been previously described in Ma et al.12 Briefly, the collection of 256 pFUS_B4 containing all possible 4-RVD combinations was synthesized by mixing all NI, HD, NN or NG RVD-containing plasmids for positions 1 through 4 (16 plasmids total) in a single Golden Gate reaction. Using this method, ∼80% of the possible combinations were identified through colony screening. The remaining pFUS_B4 clones that were not identified in the initial screen were synthesized individually. The library of 256 pFUS_B4 plasmids is available through Addgene (https://www.addgene.org/Stephen_Ekker/).
Cell culture
The HMCLs ALMC-2 and KAS-6/1 were established in our laboratory and described elsewhere.14, 15 The cells are routinely maintained in IMDM medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal calf serum and 1 ng/ml recombinant IL-6.
Transfection, selection and subcloning
A total of 20 μg of TALEN construct (left arm and right arm) and pEGFP-N1 (Clontech, Mountain View, CA, USA) DNAs were transiently cotransfected at the ratio of 2:2:1, respectively, by electroporation into 5 × 106 ALMC-2 and KAS-6/1 HMCLs. Forty-eight hours after transfection, green fluorescent protein (GFP)-expressing cells were selected by fluorescence-activated cell sorting sorting on a BD FACS Aria II sorter (BD, San Jose, CA, USA). The resulting GFP (and TALEN)-positive cells were expanded in culture for 4 additional days, followed by single-cell subcloning using limiting dilution (20 cells/96 well plate; Figure 2b).
Detection of TALEN-mediated HPRT1 disruption
To detect TALEN-mediated disruption of the HPRT1 gene, primers flanking the TALEN target site were designed using Primer3Plus (www.primer3plus.com). The primer sequences were HPRT1_F1 5′-CAGCCTCAACATCCTGCACT-3′ and HPRT1_R1 5′-CACACAATAGCTCTTCAGTCTG-3′ (schematic depicted in Figure 2a). Genomic DNA was extracted from single-cell HMCL clones, amplified with the HPRT1 primers and the resulting products were subcloned into the pCR-II vector using the TOPO TA cloning kit (Life Technologies) and sequenced with the M13 forward primer.
Sensitivity of HPRT1 disrupted cells to 6-thioguanine killing
Two independent TALEN-induced HPRT1 mutant subclones (S1 and S4) of ALMC-2 possessing a 192-nucleotide (nt) insertion and 2-nt deletion, respectively, along with the parental ALMC-2 cells were treated with increasing amounts (0, 1.0, 2.5, 5.0 and 10 μM) of 6-thioguanine (6-TG) in IMDM medium supplemented with 10% fetal calf serum, and 1 ng/ml recombinant IL-6. Cells were seeded at the density of 0.5 × 106/ml in a 24-well/plate in quadruplicate. After 9 days of culturing, cell viability in each well was assessed using a Vi-Cell XR (Beckman-Coulter, Brea, CA, USA) cell counter.
Results
To test whether TALENs are effective in HMCLs, we engineered a TALEN pair to target the HPRT1 gene as depicted in Figure 2a. The HPRT1 gene is located on the X-chromosome and encodes the protein hypoxanthine phosphoribosyltransferase that is necessary for the generation of purine nucleotides through the purine salvage pathway.16 Both male and female somatic cells have only a single functional copy of the HPRT1 gene because the second copy of the gene in female cells is transcriptionally silenced.17 The inactivation of the functional copy of the HPRT1 gene renders cells resistant to 6-TG, making it an ideal selection marker for cells in culture.17 To enrich for transfected cells, we cotransfected cells with an enhanced GFP (EGFP) expression plasmid that would allow us to use FACS to enrich for highly transfected cells that would have likely received the HPRT1 TALEN expression constructs as well. Thus, 48 h after transfection of ALMC-2 and KAS-6/1 HMCLs with the TALEN pair and pEGFP-N1 plasmid DNAs, GFP-expressing cells were isolated, allowed to rest for 2–4 days and then plated as single cells. After 2 weeks of expansion in culture, we were successful in obtaining 13 ALMC-2 and 10 KAS-6/1 healthy growing subclones.
We then examined whether the genomic sequence at the HPRT1 locus in these single-cell subclones was altered by the TALENs. Amplification of the unmodified HPRT1 gene using the primers described above should yield a 408-bp amplicon. As shown in Figure 3a, all 13 ALMC-2-derived subclones yielded a single sharp DNA band with the size of either ∼400 or 600 bp on an agarose gel, whereas most of the KAS-6/1-derived subclones (no. 3–9) produced two major products with sizes of ∼700 and 400 bp. Both cell lines were derived from female patients suggesting each cell line should have two copies of the X-chromosome, hence two TALEN targetable copies of the HPRT1 gene. This apparent difference in HPRT1 gene targeting by TALENs between ALMC-2 and KAS-6/1 MM cells prompted us to investigate the copy number of the HPRT1 gene given the fact that MM cells typically display abnormal ploidy. Indeed, prior array comparative genomic hybridization analysis of both cell lines had revealed that the ALMC-2 cell line had lost a copy of the X-chromosome, whereas the KAS-6/1 cell line retained both intact X-chromosomes (unpublished data). Therefore, the two amplified bands observed in the KAS-6/1 cell line may suggest that some of the KAS-6/1 subclones were modified at one allele while the others had identical HPRT1 mutations, or both alleles were differentially repaired resulting in distinct modifications in a single cell (Figure 3a).

Sequence analysis of TALEN-modified HPRT1 gene loci. (a) Gel images for PCR amplicons of TALEN-modified ALMC-2 and KAS-6/1 subclones. (b) Sequence analysis of representative subclones from ALMC-2 as well as KAS-6/1 cells. Sequences in red front represent TALEN binding. Areas marked in yellow are deletions, whereas the blue box represents an insertion. Two HPRT1 alleles in KAS-6/1 subclones are typically rearranged differently. S, single clone subset; Wt, wild type.
We next examined the DNA nucleotide sequences of these PCR products. Because TALEN technology can disrupt gene function via the insertion or deletion of nucleotides by error-prone non-homologous end-joining, we subcloned the DNA fragments into the TOPO TA pCR-II vector, followed by sequencing of 6–8 DNA clones for each single-cell subclone. In all, 78 bacterial colonies derived from the TOPO-cloned HPRT1 PCR products from the 13 distinct ALMC-2 subclones showed evidence of TALEN-induced modifications. Further, sequence alignment analysis showed that only two types of sequence modifications occurred. The lower band with the size of 400 bp showed a 2-nt deletion at the TALEN target site, whereas the larger band of ∼600 bp in size had a 192-nt insertion. We further searched the origin of the inserted 192-nt sequence and found it matched to a segment of the f1 replication origin sequence present in most plasmids including pC-GoldyTALEN and pEGFP-N1 used in our transfection. We also subcloned the DNA bands from TALEN-modified KAS-6/1 subclones 2, 3, 5 and 9. Although there were multiple amplification products in single-cell subclones 3 and 9, only the 400 bp band matched the HPRT1 sequence. Several bacterial colonies from each KAS-6/1 subclone were picked and those containing a 400 bp insert were sequenced. In total, 52 of these colonies were sequenced and no wild-type HPRT1 sequences were found. Furthermore, both HPRT1 alleles in each of the four cell subclones examined had different mutations (Figure 3b), suggesting that the modification of each allele is autonomous. These data clearly demonstrate that TALENs are a very robust genome-editing tool for HMCLs.
After further analysis of TALEN-modified sequences from ALMC-2 subclones S1 and S4, we found both types of HPRT1 disruption resulted in frameshifting of the HPRT1 gene open reading frame (Figure 4a). This suggests that both types of mutation would render the subclones resistant to 6-TG selection. We therefore treated the S1 subclone with the 192-nt insertion and the S4 subclone with the 2-nt deletion along with unmodified parental ALMC-2 cells with increasing concentrations of 6-TG for 9 days. As shown in Figure 4b, although parental ALMC-2 cells were readily killed by 10 μM of 6-TG, both subclones were completely resistant to the drug. Our results therefore clearly suggest that TALEN technology is indeed a very robust system for editing the MM genome. Our data also demonstrate that GFP cotransfection coupled with FACS sorting is a very efficient way to select TALEN-modified cells in a very short period of time (Figure 2b).

Functional disruption of the HPRT1 gene in ALMC-2 subclones S1 and S4. (a) Protein-coding sequence alignment shows frameshifts caused by TALENs. (b) Comparison of 6-TG sensitivity of parental ALMC-2 cells with two subclones with HPRT1 gene disruption by TALEN technology.
Discussion
The newly emerged TALEN technology has shown enormous flexibility and power in editing genomes in various model systems including Caenorhabditis elegans,18 zebrafish10, 19, 20 and embryonic stem cells21, 22, 23 with great success. Chromosomal translocations and complex gene rearrangements are common characteristics of human cancer, and have not been successfully recreated in vivo due to the lack of specific targeting tools. However, Piganeau et al.24 successfully used specific TALENs to create t(11;22)(q24;q12) and t(2;5)(p23;q35) translocations found in Ewing sarcoma in human mesenchymal precursor cells, whereas Nyquist et al.25 used TALENs to engineer prostate-specific androgen receptor gene rearrangements. Here we show that TALENs are also efficient in targeting genes in HMCLs, providing a powerful tool to dissect existing somatic mutations in MM cells. We believe the method is also broadly applicable to other cancer cells, thereby providing a great tool for the functional study of cancer mutations in general.
Among the sequences analyzed, we found that several ALMC-2 cell-derived subclones carry a 192-nt DNA insert of plasmid DNA origin. However, it has previously been reported that exogenous DNA fragments have the tendency to be integrated into the double-stranded DNA break sites of host genomes.26, 27, 28, 29 To determine whether the insert DNA was generated by TALEN off-target effects in the plasmid DNA, we examined the DNA plasmid sequences flanking the insert. However, we did not find any significant sequence homology that would qualify them as off-target sites for the HPRT1-specific TALENs. Within the exogenous insert and flanking genomic sequence in HPRT1, we found 6-base and 3-base microhomology that may have been exploited for microhomology-mediated end joining. Our observations are consistent with the findings of Lin and Waldman who found that only minimal 2 or 3 nt homologies at the junctions is needed to allow exogenous DNA insertion into host DNA at their ends.27 Such an efficient ‘absorption’ of foreign DNA into double-stranded DNA break sites actually achieves efficient disruption of the targeting gene, but may also be undesirable for other applications such as gene correction. In vitro-transcribed TALEN mRNA30 has been used with considerable success and could be used instead of plasmid DNA to avoid unwanted insertions and episomal effects.
As mentioned above, MM is known to evolve from the premalignant stage, monoclonal gammopathy of undetermined significance. Next-generation sequencing of primary patient MM cells has revealed a wide range of recurrently mutated genes, including TP53, KRAS, NRAS, BRAF, TRAF3, PRDM1, FAM46C, DIS3, ACTG1, FGFR3 and so on.3, 31, 32, 33 However, the precise pathogenic mechanism of some of these mutations and whether or not they underlie malignant progression is not clear. Our current study suggests that TALEN technology will enable us to functionally dissect these mutations in HMCLs.
References
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–339.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505: 495–501.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014; 25: 91–101.
Meacham CE, Morrison SJ . Tumour heterogeneity and cancer cell plasticity. Nature 2013; 501: 328–337.
Roebroek AJ, Wu X, Bram RJ . Knockin approaches. Methods Mol Biol 2003; 209: 187–200.
Sedivy JM, Dutriaux A . Gene targeting and somatic cell genetics–a rebirth or a coming of age? Trends Genet 1999; 15: 88–90.
Rago C, Vogelstein B, Bunz F . Genetic knockouts and knockins in human somatic cells. Nat Protoc 2007; 2: 2734–2746.
Campbell JM, Hartjes KA, Nelson TJ, Xu X, Ekker SC . New and TALENted genome engineering toolbox. Circ Res 2013; 113: 571–587.
Boch J . TALEs of genome targeting. Nat Biotechnol 2011; 29: 135–136.
Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG 2nd et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012; 491: 114–118.
Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci USA 2008; 105: 5809–5814.
Ma AC, Lee HB, Clark KJ, Ekker SC . High efficiency In Vivo genome engineering with a simplified 15-RVD GoldyTALEN design. PLoS One 2013; 8: e65259.
Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M et al. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci USA 2012; 109: 17382–17387.
Westendorf JJ, Ahmann GJ, Greipp PR, Witzig TE, Lust JA, Jelinek DF . Establishment and characterization of three myeloma cell lines that demonstrate variable cytokine responses and abilities to produce autocrine interleukin-6. Leukemia 1996; 10: 866–876.
Arendt BK, Ramirez-Alvarado M, Sikkink LA, Keats JJ, Ahmann GJ, Dispenzieri A et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood 2008; 112: 1931–1941.
Keebaugh AC, Sullivan RT, Thomas JW . Gene duplication and inactivation in the HPRT gene family. Genomics 2007; 89: 134–142.
Dobrovolsky VN, Shaddock JG, Mittelstaedt RA, Miura D, Heflich RH . Detection of in vivo mutation in the Hprt and Pig-a genes of rat lymphocytes. Methods Mol Biol 2013; 1044: 79–95.
Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011; 333: 307.
Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B . Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 2011; 29: 699–700.
Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK et al. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol 2011; 29: 697–698.
Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 2011; 29: 695–696.
Liu H, Chen Y, Niu Y, Zhang K, Kang Y, Ge W et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 2014; 14: 323–328.
Frank S, Skryabin BV, Greber B . A modified TALEN-based system for robust generation of knock-out human pluripotent stem cell lines and disease models. BMC Genomics 2013; 14: 773.
Piganeau M, Ghezraoui H, De Cian A, Guittat L, Tomishima M, Perrouault L et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res 2013; 23: 1182–1193.
Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA et al. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci USA 2013; 110: 17492–17497.
Lin Y, Waldman AS . Promiscuous patching of broken chromosomes in mammalian cells with extrachromosomal DNA. Nucleic Acids Res 2001; 29: 3975–3981.
Lin Y, Waldman AS . Capture of DNA sequences at double-strand breaks in mammalian chromosomes. Genetics 2001; 158: 1665–1674.
Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 2011; 29: 816–823.
Miller DG, Petek LM, Russell DW . Adeno-associated virus vectors integrate at chromosome breakage sites. Nat Genet 2004; 36: 767–773.
Li C, Qi R, Singleterry R, Hyle J, Balch A, Li X et al. Simultaneous Gene Editing by Injection of mRNAs Encoding Transcription Activator-Like Effector Nucleases (TALENS) into Mouse Zygotes. Mol Cell Biol 2014; 34: 1649–1658.
Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471: 467–472.
Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012; 120: 1060–1066.
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014; 5: 2997.
Acknowledgements
This article was supported by the State of Minnesota for the University of Minnesota Mayo Clinic Partnership Grant H001274506, NIH Grant GM63904, NIDDK P30DK084567 to SCE and CA164232 to DFJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Wu, X., Blackburn, P., Tschumper, R. et al. TALEN-mediated genetic tailoring as a tool to analyze the function of acquired mutations in multiple myeloma cells. Blood Cancer Journal 4, e210 (2014). https://doi.org/10.1038/bcj.2014.32
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2014.32
