
Abstract
Aberrant activation of Janus kinase 2 (JAK2) caused by somatic mutation of JAK2 (JAK2V617F) or the thrombopoietin receptor (MPLW515L) plays an essential role in the pathogenesis of myeloproliferative neoplasms (MPNs), suggesting that inhibition of aberrant JAK2 activation would have a therapeutic benefit. Our novel JAK2 inhibitor, NS-018, was highly active against JAK2 with a 50% inhibition (IC50) of <1 nM, and had 30–50-fold greater selectivity for JAK2 over other JAK-family kinases, such as JAK1, JAK3 and tyrosine kinase 2. In addition to JAK2, NS-018 inhibited Src-family kinases. NS-018 showed potent antiproliferative activity against cell lines expressing a constitutively activated JAK2 (the JAK2V617F or MPLW515L mutations or the TEL–JAK2 fusion gene; IC50=11–120 nM), but showed only minimal cytotoxicity against most other hematopoietic cell lines without a constitutively activated JAK2. Furthermore, NS-018 preferentially suppressed in vitro erythropoietin-independent endogenous colony formation from polycythemia vera patients. NS-018 also markedly reduced splenomegaly and prolonged the survival of mice inoculated with Ba/F3 cells harboring JAK2V617F. In addition, NS-018 significantly reduced leukocytosis, hepatosplenomegaly and extramedullary hematopoiesis, improved nutritional status, and prolonged survival in JAK2V617F transgenic mice. These results suggest that NS-018 will be a promising candidate for the treatment of MPNs.
Similar content being viewed by others


Introduction
The breakpoint cluster region-abelson (BCR-ABL)-negative myeloproliferative neoplasms (MPNs) include polycythemia vera (PV), essential thrombocythemia and primary myelofibrosis.1, 2 Current treatment options for MPNs, and especially for primary myelofibrosis, are limited and largely palliative with the notable exception of allogeneic stem cell transplantation.3 A somatic point mutation at codon 617 of Janus kinase 2 (JAK2) tyrosine kinase (TYK) (JAK2V617F) has been shown to occur in ∼95% of patients with PV and ∼50% of patients with essential thrombocythemia and primary myelofibrosis.4, 5, 6, 7, 8 JAK2V617F is a constitutively activated kinase that activates the JAK/STAT signaling pathway and dysregulates cell growth and function. Expression of JAK2V617F transforms hematopoietic cells to cytokine-independent growth in vitro and causes MPN-like diseases in mice after bone marrow transplantation.5, 9, 10, 11, 12 Transgenic mice expressing JAK2V617F also develop MPN-like diseases.13, 14, 15, 16, 17, 18 In addition, other somatic mutations leading to aberrant JAK2 activation, that is, activating mutations in exon 12 of JAK2 and mutations at codon 515 of the thrombopoietin receptor (MPLW515L/K), have been identified in JAK2V617F-negative MPN patients.19, 20 These findings suggest that the inhibition of aberrant JAK2 activation would have a therapeutic benefit, and several JAK2 inhibitors are currently in clinical trials for patients with MPNs.21, 22
NS-018 is a newly discovered, orally bioavailable, small-molecule inhibitor of JAK2 that is competitive with adenosine triphosphate (ATP). In this study, we describe the preclinical characterization of NS-018 and report on its potent and selective inhibitory activity against JAK2 and Src-family kinases and promising in vitro and in vivo activity against constitutively active JAK2 mutants.
Materials and methods
Structural analysis
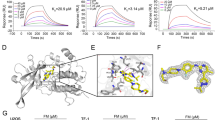
The kinase domain of human JAK2 was expressed in Sf9 cells infected with recombinant virus and purified as described elsewhere.23 The NS-018/protein complex was concentrated and crystallized by the hanging drop method at 4 °C. Diffraction data from flash-frozen crystals were collected at the BL32B2 beamline of the SPring-8 synchrotron facility (Hyogo, Japan) and processed with the HKL-2000 package.24 The structure was solved by molecular replacement with the program Phaser.25 All computations were performed with Molecular Operating Environment version 2009.10 (Chemical Computing Group, Montreal, QC, Canada). Figure 1 was prepared with PyMOL version 1.3 (Schrödinger, New York, NY, USA).

X-ray co-crystal structure of NS-018 bound to JAK2 kinase. The surface of NS-018 is shown in pink, the backbone of JAK2 in slate, and the A-loop in cyan. Gly993 is located immediately N-terminal to the DFG motif. For clarity, only key residues are shown.
In vitro kinase assay
The kinase domains of human JAK1, JAK2, JAK3 and TYK2 were purchased from Carna Biosciences (Kobe, Japan). Each kinase was incubated in a reaction medium containing serial dilutions of NS-018, biotinylated peptide substrate, ATP and MgCl2 in a streptavidin-coated plate for 1 h at 30 °C. Phosphorylated substrates were spectrophotometrically detected with horseradish peroxidase-linked antibody (PY-20; BD Biosciences, San Jose, CA, USA) and TMB (3,3′,5,5′-tetramethylbenzidine) solution (Sigma Aldrich, St Louis, MO, USA). The concentrations required to give 50% inhibition (IC50) were estimated by fitting the absorbance data to a logistic curve with SAS version 8.2 (SAS Institute, Cary, NC, USA). The inhibitory effect of NS-018 was tested against a panel of 53 kinases by Carna Biosciences according to their internal protocol.
Cellular assay
Cell lines were used after reaching 70–90% confluence. For cell growth assay, cells were seeded in 96-well plates at densities optimized for growth rate (transformed Ba/F3 cell lines at 1 × 103 cells/well, SET-2 cells at 1 × 104 cells/well, MV4-11 cells at 2 × 104 cells/well and other cell lines at 5 × 103 cells/well). The next day, cells were treated with serial dilutions of NS-018, and incubated for 72 h at 37 °C with 5% CO2. Viability was measured by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. IC50 values were estimated with SAS version 8.2. For western blotting and apoptosis, see Supplementary Materials and methods.
Colony formation assay
Peripheral blood mononuclear cells from PV patients with the JAK2V617F mutation or healthy volunteers were collected with informed consent and Institutional Review Board approval. A total of 2 × 105 cells were treated with increasing concentrations of NS-018 in MethoCult H4534 methylcellulose medium (StemCell Technologies, Vancouver, BC, Canada) supplemented with or without 3 U/mL erythropoietin. Experiments were performed in triplicate. Burst-forming unit-erythroids were counted on day 14. IC50 values were estimated with SAS version 8.2.
Mouse Ba/F3-JAK2V617F disease model
The study was conducted in compliance with the Law for the Humane Treatment and Management of Animals (Law No. 105, 1973, as revised on 1 June 2006). Female BALB/c nude mice (Japan SLC, Shizuoka, Japan) were placed in blanket cages in an environment maintained at 21–25 °C and 45–65% relative humidity, with artificial illumination for 12 h and a ventilation frequency of at least 15 times/h. They were allowed free access to food pellets and tap water. Ba/F3-JAK2V617F cells (106 per mouse) were inoculated intravenously into 7-week-old mice. Administration of vehicle (0.5% methylcellulose) or NS-018 twice daily by oral gavage began the day after cell inoculation. Survival was monitored daily, and moribund mice were humanely killed and their time of death was recorded for purposes of survival analysis. In a parallel study, all mice were humanely killed after 8 days of administration, and their spleens were removed and weighed.
JAK2V617F transgenic mice
The generation and genotyping of transgenic mice (BDF1 strain) were carried out as described previously.15 At 12 weeks after birth, treatment with vehicle or NS-018 was begun by oral gavage and was continued twice a day on weekdays for 24 weeks. The body weight of the mice was measured weekly. Peripheral blood was drawn monthly into heparin-coated glass capillary tubes, and hematological parameters were determined with a Celltac-α hematology analyzer (Nihon Kohden, Tokyo, Japan). For fractional analysis of white blood cells (WBCs), nucleated cells were stained with fluorescently conjugated antibodies specific for Mac1, Gr1, B220 and CD3 (BD Biosciences) and the percentage of each fraction was determined with a FACSCalibur (BD Biosciences). The fractional cell number was computed by multiplying the percentage by the total WBC count.
All mice were humanely killed at the end of treatment, and terminal blood samples and organs were collected. For flow cytometric analysis of spleen and bone marrow, see Supplementary Materials and methods. For histological evaluation, tissues samples from liver, spleen, lung and femur were fixed in formalin, embedded in paraffin and cut for hematoxylin-eosin staining or Gomori silver staining according to standard protocols (Biopathology Institute, Oita, Japan). Histological slides were viewed under a BX50 microscope and photographed with a FX380 digital camera (Olympus, Tokyo, Japan).
Results
NS-018 is a potent and selective JAK2 kinase inhibitor in vitro
NS-018 was discovered by screening for potent and selective JAK2 inhibitors. Structure-activity studies leading to the identification of NS-018 as a promising candidate and a description of its synthesis will be published elsewhere. In in vitro kinase assays, NS-018 was highly active against JAK2 with an IC50 of 0.72 nM, and it had 30–50-fold greater selectivity for JAK2 over other Jak-family kinases such as JAK1, JAK3 and TYK 2 (Table 1 ). The ability of NS-018 to inhibit other kinases was tested with a panel of 53 kinases in the presence of ATP at concentrations close to their respective Km values. NS-018 showed potent inhibition of Src-family kinases, notably SRC and FYN, as well as weak inhibition of ABL and FLT3 with 45- and 90-fold selectivity for JAK2, respectively (Table 2 ). NS-018 showed a high degree of selectivity for JAK2 over many other TYKs (Table 2) and serine/threonine kinases (Supplementary Table 1).
Structural analysis of JAK2 kinase in complex with NS-018
Phosphorylation of the activation loop (A-loop) is one of the most common mechanisms for regulating protein kinase activity, and it leads to the relocation of an Asp–Phe–Gly (DFG) motif located adjacent to the N-terminus of the A-loop.26 The X-ray co-crystal structure of JAK2 in complex with NS-018 revealed that (1) Tyr residues at positions 1007 and 1008 in the A-loop were phosphorylated, (2) the phosphorylated A-loop lay outside the active-site cleft and (3) NS-018 bound to JAK2 in the ‘DFG-in’ active conformation (Figure 1).
NS-018 inhibits JAK2-mediated signaling and proliferation and induces apoptosis
To evaluate the effects of NS-018 on JAK2-mediated signaling, we exposed Ba/F3 cells expressing JAK2V617F (Ba/F3-JAK2V617F) to increasing concentrations of NS-018 for 3 h and measured the level of phosphorylation of JAK2-mediated signaling components by western blotting (Figure 2a). NS-018 inhibited the phosphorylation of STAT5, STAT3 and ERK in a dose-dependent manner, with maximal effects at ∼100 nM, 30 nM and 300 nM, respectively.

Inhibition of JAK2-mediated signaling and induction of apoptosis in Ba/F3-JAK2V617F cells by NS-018. (a) Ba/F3-JAK2V617F cells were treated with increasing concentrations of NS-018 for 3 h and then lysed. Lysate was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a Polyvinylidene fluoride membrane and probed with the indicated antibodies. (b) Ba/F3-JAK2V617F cells were treated with 0–300 nM NS-018 for 48 h and tested for apoptosis by staining with annexin V/propidium iodide (PI). (c) Ba/F3-JAK2V617F cells were treated with NS-018 for 29 h before DNA extraction and DNA fragmentation was assessed by agarose gel electrophoresis. M indicates the DNA size marker.
We next assessed the antiproliferative activity of NS-018 against hematopoietic cell lines (Table 3 ). NS-018 suppressed the growth of Ba/F3-JAK2V617F cells with an IC50 of 60 nM and the JAK2V617F-positive cell line SET-2 with an IC50 of 120 nM. NS-018 also inhibited the growth of Ba/F3-MPLW515L cells, which is dependent on JAK2-mediated signaling due to an activating mutation of the thrombopoietin receptor, at similar concentrations. Ba/F3-TEL-JAK2 cells were highly sensitive to NS-018, but Ba/F3-TEL-JAK3 cells were less sensitive. CMK cells, which are dependent on both JAK1 and JAK3 because of an activating mutation of JAK3 that signals through wild-type JAK1,27 were also insensitive to NS-018. NS-018 showed weak antiproliferative activity against K-562 cells, which carry BCR-ABL and MV4-11 cells, which carry an internal tandem duplication of FLT3. This selective antiproliferative activity was roughly consistent with the kinase inhibitory profile of NS-018. Additionally, NS-018 showed only minimal cytotoxicity against other hematopoietic cell lines, such as SKM-1 and U-937.
To determine whether the antiproliferative activity of NS-018 was accompanied by an increase in apoptosis, we exposed Ba/F3-JAK2V617F cells to various concentrations of NS-018 and determined the percentages of apoptotic cells by flow cytometry with annexin V/propidium iodide staining and assessed DNA fragmentation. NS-018 treatment increased both the percentage of annexin V-positive cells (Figure 2b) and the extent of DNA fragmentation (Figure 2c) in a dose-dependent manner. Thus, NS-018 both inhibited the phosphorylation of components of JAK2-mediated signaling and induced apoptosis in cell lines whose growth depended on JAK2 activation.
NS-018 inhibits erythroid progenitor cell growth in primary PV patient samples
To evaluate the efficacy of NS-018 against primary MPN patient cells, we performed colony formation assays with mononuclear cells from the peripheral blood of PV patients with the JAK2V617F mutation or of healthy volunteers. NS-018 inhibited the formation of burst-forming unit-erythroid from healthy controls and PV patients in a dose-dependent manner, but the degree of inhibition was significantly greater for the PV patients (P=0.011). Specifically, for three healthy controls, NS-018 inhibited erythroid colony growth with a mean IC50 of 952±118 nM, whereas for four PV patients the corresponding IC50 was 529±36 nM (Figure 3a). We also assessed the efficacy of NS-018 in inhibiting the growth of erythropoietin-independent, endogenous erythroid colony formation, a hallmark of JAK2V617F-positive MPN. NS-018 inhibited endogenous erythroid colony formation with a mean IC50 of 224±26 nM (Figure 3b). Thus, NS-018 effectively inhibited erythroid progenitor cell growth in PV patient samples.

Effect of NS-018 on erythroid colony formation by PV patient cells. Mononuclear cells isolated from the peripheral blood of PV patients with the JAK2V617F mutation (n=4) or healthy volunteers (n=3) were treated with increasing concentrations of NS-018 in methylcellulose-based media with erythropoietin (a) or without erythropoietin (b). BFU-Es were counted on day 14. Values represent the mean±s.e.m. Statistical significance was assessed by the t-test (*P<0.05).
NS-018 is effective in a mouse Ba/F3-JAK2V617F disease model
We next evaluated the in vivo efficacy of NS-018 in a mouse acute disease model. Mice inoculated with Ba/F3-JAK2V617F cells showed marked splenomegaly and died within 2–3 weeks because of penetrant hematopoietic disease progression. NS-018, administered by oral gavage twice a day, significantly prolonged survival of the mice at dosages of 12.5 mg/kg or higher (Figure 4a). Although vehicle-treated mice had all died by day 19, all mice treated with 100 mg/kg NS-018 were still alive even on day 25. NS-018 also significantly reduced splenomegaly at dosages of 1.5 mg/kg or higher (Figure 4b). The weight and appearance of the spleens of mice treated with 50 mg/kg NS-018 were similar to those of uninoculated control mice. Thus, NS-018 was highly efficacious in this mouse model of acute disease.

Effect of NS-018 on survival and splenomegaly in a mouse Ba/F3-JAK2V617F disease model. Disease was established in BALB/c nude mice by intravenous injection of 1 × 106 Ba/F3-JAK2V617F cells. (a) Survival curves. NS-018 was administered by oral gavage twice a day for 11 days at the indicated doses. Statistical significance was assessed by the log-rank test (*P<0.01 vs vehicle, eight mice per group). (b) Mice inoculated with Ba/F3-JAK2V617F were orally administered with the indicated doses of NS-018 twice a day. Uninoculated control mice (sham) received vehicle only. The day after the completion of 8 days of administration, spleens were removed and weighed. Values represent the mean±s.e.m. Statistical significance was assessed by the t-test (#P<0.01 vs sham) and Dunnett's test (*P<0.01 vs vehicle, five mice per group).
Efficacy of NS-018 in mouse MPN model (JAK2V617F transgenic mice)
Mice expressing JAK2V617F under the control of the H2Kb promoter (V617F-transgenic mice (TG)) show an MPN phenotype, including leukocytosis, thrombocytosis, progressive anemia, hepatosplenomegaly with extramedullary hematopoiesis, megakaryocyte hyperplasia and fibrosis in the bone marrow.15 They also exhibit body weight loss and high mortality compared with wild-type controls. Their bone marrow cells show constitutive activation of STAT5 and cytokine-independent erythroid colony formation. In this study, we tested the efficacy of NS-018 in this chronic MPN model. Before starting long-term administration, we confirmed that NS-018 inhibited constitutive JAK2-mediated signaling in vivo. As expected, NS-018 inhibited the phosphorylation of STAT5 in Mac1+/Gr1+ myeloid cells from bone marrow of V617F-TG mice following a single oral administration at a dosage of 50 mg/kg (Supplementary Figure 1).
After disease was established at 12 weeks after birth, V617F-TG mice were randomly assigned to treatment with NS-018 or vehicle. NS-018 was administered by oral gavage twice a day for 24 weeks at doses of 25 or 50 mg/kg, and the control groups received vehicle only. No signs of gross toxicity were observed during the 24 weeks of treatment.
During the study, the peripheral blood count of the mice was monitored monthly (Figures 5A–D). V617F-TG mice showed marked leukocytosis. After 4 weeks of NS-018 treatment, the WBC count was reduced to 59% in the 25 mg/kg per group and 39% in the 50 mg/kg per group compared with the vehicle-treated group, and the effect was maintained until the end of the study (Figure 5A). To determine which kinds of WBC increased, we performed a fractional analysis by flow cytometry. At 8 weeks, the numbers of Mac1+/Gr1+ myeloid cells, B220+ B cells and CD3+ T-cells in V617F-TG mice were respectively 370-, 5.4- and 8.8-fold greater than in wild-type (WT) mice (Figure 5B). In the 50 mg/kg per group, the respective numbers fell to 98-, 3.3- and 5.3-fold. Although NS-018 reduced the numbers of all WBC types, the reduction in Mac1+/Gr1+ myeloid cells was the greatest. V617F-TG mice also showed progressive anemia (Figure 5C). The 25 mg/kg per group followed the same course of reduction in red blood cell count as the vehicle-treated group. However, the 50 mg/kg per group showed no reduction in red blood cell count even after 20 weeks, although the count was lower than that of WT mice. This indicated that treatment with 50 mg/kg NS-018 prevented the progression of anemia. V617F-TG mice showed thrombocytosis in the early stages, but the platelet (PLT) count declined with time (Figure 5D). PLT aggregation and giant PLTs were observed in the peripheral blood of these mice.15 NS-018 treatment resulted in sustained thrombocytosis.


Effect of NS-018 in JAK2V617F transgenic mice. JAK2V617F transgenic mice (TG) were divided into three groups and orally administered with vehicle, 25 mg/kg NS-018, or 50 mg/kg NS-018 (n=34, 37, and 36, respectively) twice a day for 24 weeks. Wild-type mice (WT) received vehicle only (n=27). (A) White blood cell (WBC), (C) red blood cell (RBC) and (D) platelet (PLT) counts were monitored monthly. Values represent the mean±s.e.m. Statistical significance was assessed by the t-test (*P<0.05, **P<0.01 vs TG vehicle). (B) Differences in cellular composition. Nucleated cells from peripheral blood were stained with fluorescently conjugated antibodies specific for Mac1/Gr1 (myeloid cells), B220 (B cells) and CD3 (T-cells) and analyzed by flow cytometry. (E) The weight of liver and spleen were measured at the end of the treatment period. Values represent the mean±s.e.m. Statistical significance was assessed by the t-test (#P<0.01 vs WT vehicle) or Dunnett's test (*P<0.05, **P<0.01 vs TG vehicle). (F) Spleen cells were stained with fluorescently conjugated antibodies specific for Mac1/Gr1 (myeloid cells), B220 (B cells) and CD3 (T-cells) and analyzed by flow cytometry. (G) After NS-018 treatment, tissue sections were prepared from spleen (a–d; original magnification × 40), liver (e–h; magnification × 200) and lung (i–l; magnification × 200) for staining with hematoxylin and eosin. (H) The body weight of the mice was monitored weekly. (I) The total cholesterol in the serum at the end of the study. Values represent the mean±s.e.m. Statistical significance was assessed by the t-test (#P<0.01 vs WT vehicle) or Dunnett's test (*P<0.01 vs TG vehicle). (J) Survival curves of mice. Statistical significance was assessed by the log-rank test (#P<0.01 vs WT vehicle, *P<0.01 vs TG vehicle).
NS-018 treatment also reduced hepatosplenomegaly in a dose-dependent manner (Figure 5E). In the spleen, NS-018 treatment significantly decreased Mac1+/Gr1+ myeloid cells associated with extramedullary hematopoiesis and significantly increased B220+ B cells (Figure 5F). Consistent with the reduction in organ weights and infiltrating myeloid cells, the histopathological results showed that NS-018 markedly reduced cell invasion and restored normal architecture (Figure 5G). In the spleen of V617F-TG mice, the white pulp was blended throughout and partially preserved, and the red pulp was expanded by mainly myeloid cell invasion. However, NS-018 treatment resulted in a marked reduction in cell invasion and a restored architecture of the spleen (Figures 5Ga–d). In the liver and lung, extramedullary hematopoiesis consisting of myeloid cells and megakaryocytes was reduced in a dose-dependent manner (Figures 5Ge–l) and almost completely disappeared in the 50 mg/kg per group (Figures 5Gh and l). In contrast to the pathological improvement in these organs, NS-018 treatment had little impact on the progression of fibrosis and megakaryocyte hyperplasia in the bone marrow.
NS-018 improves survival and compromised nutritional status in a mouse MPN model
V617F-TG mice exhibited reduced body weight gain and high mortality compared with wild-type controls (Figures 5H and J). However, mice treated with 50 mg/kg NS-018 progressively gained more weight than vehicle-treated mice, and their body weight was comparable to that of WT mice (Figure 5H). Total cholesterol was significantly decreased in the serum of V617F-TG mice compared with WT mice, indicating compromised nutritional status. However, in accordance with the body weight gain, the total cholesterol was increased in the NS-018-treated groups at the end of the study (Figure 5I). NS-018 also improved the survival of V617F-TG mice. During the 24-week study, 12 of 34 mice died in the vehicle group, whereas 1 of 36 mice died in the 50 mg/kg per group (Figure 5J). This corresponds to a statistically significant prolongation of survival in the 50 mg/kg per group (P<0.01). Taken together, these results suggest that NS-018 reduced leukocytosis, anemia progression, hepatosplenomegaly and extramedullary hematopoiesis, improved nutritional status, and prolonged survival in V617F-TG mice.
Discussion
In view of the lack of satisfying treatment options for patients with BCR-ABL-negative MPNs, we sought to develop an orally bioavailable small-molecule therapeutic agent to treat these diseases. The discovery of the JAK2V617F and MPLW515L mutations in MPN patients suggests that the inhibition of aberrant JAK2 activation would have a therapeutic benefit for MPN patients. Our novel JAK2 inhibitor, NS-018, was found to be highly active against JAK2 with an IC50 of <1 nM and to have high selectivity for JAK2 over many other kinases.
In addition to JAK2, NS-018 inhibited Src-family and ABL kinases with up to almost 50-fold selectivity for JAK2 (Table 2). To investigate the structural factors determining the selectivity of NS-018, we carefully explored the binding site of the X-ray co-crystal structure of the complex of the human JAK2 kinase domain and NS-018 (Figure 1). Gly at position 993, which is located immediately N-terminal to the A-loop DFG motif, tightly fixed the position of NS-018. Because Gly is the smallest amino acid, we hypothesized that NS-018 effectively inhibites kinases with small amino acids at this position. In keeping with this hypothesis, NS-018 was active against ABL and Src-family kinases, which have Ala, the second smallest natural amino acid, at this position.28 However, kinases belonging to the Axl, FGFR, InsR, Met and Tie families have Gly or Ala at this position, yet NS-018 did not inhibit them. These kinases may have amino acids at other positions that prevent NS-018 binding. The remaining kinases listed in Table 2 have larger amino acids such as Cys, Ser or Thr at this position, and NS-018 did not inhibit these kinases. Furthermore, the Ser/Thr kinases listed in Supplementary Table 1 also have larger amino acids such as Cys, Ser, Thr, Val, Leu or Ile at the corresponding position, and NS-018 also did not inhibit these kinases. These results provide evidence that the selectivity of NS-018 is largely determined by the size of the amino acid at position 993.
JAK and Src-family kinases work in concert to activate many signaling molecules.29 Cooperation between SRC and JAKs is required for full activation of STAT3.30 Potent inhibition of STAT3 phosphorylation in Ba/F3-JAK2V617F cells by NS-018 (Figure 2a) may be explained by the simultaneous inhibition of JAK2 and Src-family kinases. Several reports have indicated the involvement of Src-family kinases in the pathogenesis of MPNs. For example, a TEL-lyn fusion gene has been identified in a patient with primary myelofibrosis.31 The Src-family kinase inhibitors dasatinib and PP2 have been shown to suppress erythropoietin-independent erythroid colony growth from PV.32, 33 Furthermore, SRC kinase preactivation is associated with PLT hypersensitivity in essential thrombocythemia and PV patient samples.34 On the other hand, LYN, FGR and HCK have been reported to be independent of JAK2V617F-induced polycythemia in a murine retroviral bone marrow transplantation model.12 Although the involvement of Src-family kinases in MPNs has not yet been fully clarified, simultaneous inhibition of JAK2 and some Src-family kinases is expected to be advantageous in preventing aberrant JAK2-STAT signaling and thereby curing the disease.
NS-018 inhibited the growth of cells, which depended on JAK2 activation with IC50 values of 11–120 nM. Consistent with the selective inhibition by NS-018 of the enzymatic activity of JAK2 over that of JAK1 and JAK3, Ba/F3-TEL-JAK3 and CMK cells were less sensitive to NS-018. Weak inhibition of ABL and FLT3 kinases by NS-018 is the most likely explanation for its weak antiproliferative activity against K-562 cells (which express BCR-ABL) and MV4-11 cells (which express an internal tandem duplication of FLT3). The difference between the selectivity of NS-018 in the enzyme-inhibition assay and in the cell growth assay may arise from a difference in the extent to which cell growth depends on kinase activation in these cell lines. The fact that NS-018 did not inhibit other Tyr or Ser/Thr kinases might explain its low general cytotoxicity against nontarget cells.
The efficacy of several JAK2 inhibitors has been evaluated in an acute mouse Ba/F3-JAK2V617F disease model.35, 36, 37 In this study, NS-018 seemed as effective as these inhibitors in this model (Figure 4). These results demonstrate the in vivo potency of NS-018. To further evaluate the efficacy of NS-018 in a chronic MPN model, we performed long-term administration of NS-018 to transgenic mice expressing JAK2V617F. In contrast to other reports that several JAK2V617F transgenic mice tend to show polycythemia,13, 14, 16, 17, 18 our transgenic mice showed progressive anemia.15 Although the reason for this is unclear, any impairments in the differentiation of erythrocyte progenitors to mature erythrocytes and the progression of bone marrow fibrosis are supposed to be related to anemia (unpublished data). Treatment with 50 mg/kg NS-018 prevented the progression of anemia in these mice (Figure 5C). To assess the causes of differences in the peripheral blood count, we examined the effects of NS-018 on hematopoietic cellular compartments and differentiation in bone marrow and spleen by flow cytometric analysis. No significant differences were observed in the proportion of hematopoietic stem cells (CD34+/Kit+/Sca1+/Lineage−), common myeloid progenitors (CD34+/Kit+/Sca1−/FcγRlow/Lineage−), granulocyte/macrophage progenitors (CD34+/Kit+/Sca1−/FcγRhigh/Lineage−), megakaryocytic/erythroid progenitors (CD34low/Kit+/Sca1−/FcγRlow/Lineage−), erythrocyte progenitors (CD71+/Ter119+) or megakaryocytic progenitors (CD41+/Kit+/CD9+/FcγRlow/Lineage−) in the NS-018-treated group compared with the vehicle-treated group (data not shown). Because the proportions of stem cells and progenitors in the spleen and bone marrow were not changed by NS-018 administration, it was assumed that erythrocyte progenitors did not increase. Additionally, serum levels of erythropoietin and thrombopoietin were not significantly different in NS-018-treated and vehicle-treated V617F-TG mice. Thus, the reason for the NS-018-dependent reduction in anemia progression remains unclear.
V617F-TG mice also showed thrombocytosis in the early stages, but the PLT count slowly decreased with time. Because the megakaryocyte number in bone marrow in these mice was remained higher than in WT mice, PLT production was assumed not to have decreased. One possible explanation for the reduction in PLT count is enhancement of PLT trapping due to progressive splenomegaly. The sustained thrombocytosis caused by NS-018 treatment (Figure 5D) was considered to be the result of reduced splenomegaly. Another possible explanation for the PLT-count reduction in V617F-TG mice is a reduced PLT life span due to the enhanced PLT aggregation. It has been hypothesized that, in patients with MPNs, continuous leukocyte degranulation due to leukocyte activation might result in the consumption of factor V and protein S, leading to activated protein C resistance and increased risk of thrombosis.38 PLT aggregation has also been observed in V617F-TG mice.15 Although NS-018 has been shown not to affect the clotting function of blood from normal rats (unpublished data), treatment with NS-018 might reduce PLT aggregation by suppressing leukocyte activation, thereby prolong PLT life span in these mice.
Loss of appetite and deterioration in nutritional status are observed in patients with MPNs, especially myelofibrosis.39 V617F-TG mice also exhibited reduced body weight gain and reduced total serum cholesterol levels, and NS-018 markedly increased both their body weight gain and their cholesterol levels (Figures 5H and I). These effects may have been brought about by an improvement in appetite through a reduction in the mechanical compression of the gastrointestinal tract resulting from hepatosplenomegaly, as well as inhibition of leptin signaling through JAK2. Improvement in nutritional status by NS-018 seemed to contribute to the prolonged survival of these mice.
In summary, NS-018 significantly reduced leukocytosis, hepatosplenomegaly and extramedullary hematopoiesis, and improved nutritional status in V617F-TG mice. All these effects of NS-018 may contribute to the survival benefits observed in V617F-TG mice. Additionally, NS-018 inhibited erythroid colony formation by peripheral blood mononuclear cells from PV patients at concentrations significantly lower than those required for healthy controls (Figure 3). Examination of the X-ray co-crystal structure showing NS-018 bound to JAK2 in the ‘DFG-in’ active conformation strongly suggests that NS-018 may inhibit constitutively activated JAK2 with the V617F mutation as well as or better than it inhibits activated wild-type JAK2. Overall, our results suggest that NS-018 will be effective in treating patients with MPNs. The efficacy and safety of NS-018 for MPNs is expected to be verified by early-phase clinical trials to begin in early 2011.
References
Campbell PJ, Green AR . The myeloproliferative disorders. N Engl J Med 2006; 355: 2452–2466.
Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007; 110: 1092–1097.
Tefferi A . Essential thrombocythemia, polycythemia vera, and myelofibrosis: current management and the prospect of targeted therapy. Am J Hematol 2008; 83: 491–497.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem 2005; 280: 22788–22792.
Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG . Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 2006; 107: 4274–4281.
Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL . JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 2006; 108: 1652–1660.
Bumm TG, Elsea C, Corbin AS, Loriaux M, Sherbenou D, Wood L et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 2006; 66: 11156–11165.
Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One 2006; 1: e18.
Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008; 111: 3931–3940.
Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood 2008; 111: 5109–5117.
Shide K, Shimoda HK, Kumano T, Karube K, Kameda T, Takenaka K et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia 2008; 22: 87–95.
Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG . Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood 2010; 115: 3589–3596.
Marty C, Lacout C, Martin A, Hasan S, Jacquot S, Birling MC et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood 2010; 116: 783–787.
Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood 2010; 116: 1528–1538.
Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356: 459–468.
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010; 363: 1117–1127.
Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol 2011; 28: 1–8.
Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood 2006; 107: 176–183.
Otwinowski Z, Minor W . Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997; 276: 307–326.
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ . Phaser crystallographic software. J Appl Cryst 2007; 40: 658–674.
Lindauer K, Loerting T, Liedl KR, Kroemer RT . Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng 2001; 14: 27–37.
Tyner JW, Walters DK, Willis SG, Luttropp M, Oost J, Loriaux M et al. RNAi screening of the tyrosine kinome identifies therapeutic targets in acute myeloid leukemia. Blood 2008; 111: 2238–2245.
Huang D, Zhou T, Lafleur K, Nevado C, Caflisch A . Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 2010; 26: 198–204.
Ingley E, Klinken SP . Cross-regulation of JAK and Src kinases. Growth Factors 2006; 24: 89–95.
Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001; 20: 2499–2513.
Tanaka H, Takeuchi M, Takeda Y, Sakai S, Abe D, Ohwada C et al. Identification of a novel TEL—Lyn fusion gene in primary myelofibrosis. Leukemia 2010; 24: 197–200.
Wappl M, Jaeger E, Streubel B, Gisslinger H, Schwarzinger I, Valent P et al. Dasatinib inhibits progenitor cell proliferation from polycythaemia vera. Eur J Clin Invest 2008; 38: 578–584.
Ugo V, Marzac C, Teyssandier I, Larbret F, Lécluse Y, Debili N et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol 2004; 32: 179–187.
Randi ML, Brunati AM, Scapin M, Frasson M, Deana R, Magrin E et al. Src tyrosine kinase preactivation is associated with platelet hypersensitivity in essential thrombocythemia and polycythemia vera. Blood 2010; 115: 667–676.
Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007; 21: 1658–1668.
Liu PCC, Caulder E, Li J, Waeltz P, Margulis A, Wynn R et al. Combined inhibition of Janus kinase 1/2 for the treatment of JAK2V617F-driven neoplasms: selective effects on mutant cells and improvements in measures of disease severity. Clin Cancer Res 2009; 15: 6891–6900.
Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010; 115: 3109–3117.
Cervantes F, Arellano-Rodrigo E, Alvarez-Larrán A . Blood cell activation in myeloproliferative neoplasms. Haematologica 2009; 94: 1484–1488.
Mishchenko E, Tefferi A . Treatment options for hydroxyurea-refractory disease complications in myeloproliferative neoplasms: JAK2 inhibitors, radiotherapy, splenectomy and transjugular intrahepatic portosystemic shunt. Eur J Haematol 2010; 85: 192–199.
Acknowledgements
We thank Dr Y Kubuki (Blood Transfusion Division, Miyazaki University), Dr K Maeda (Miyakonojo National Hospital), and Dr K Yamashita (Miyazaki Prefectural Miyazaki Hospital) for collecting the patient samples. We also thank Dr T Kameda, M Matsushita and T Shinmori for technical advice and assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Blood Cancer Journal website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Nakaya, Y., Shide, K., Niwa, T. et al. Efficacy of NS-018, a potent and selective JAK2/Src inhibitor, in primary cells and mouse models of myeloproliferative neoplasms. Blood Cancer Journal 1, e29 (2011). https://doi.org/10.1038/bcj.2011.29
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2011.29
Keywords
This article is cited by
-
JAK2 inhibitors for the treatment of Philadelphia-negative myeloproliferative neoplasms: current status and future directions
Molecular Diversity (2023)
-
The role of driver mutations in myeloproliferative neoplasms: insights from mouse models
International Journal of Hematology (2020)
-
Targeted therapies for myeloproliferative neoplasms
Biomarker Research (2019)
-
NS-018 reduces myeloma cell proliferation and suppresses osteolysis through inhibition of the JAK2 and Src signaling pathways
Blood Cancer Journal (2018)
-
A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis
Leukemia (2017)

{kind=link}