
Abstract
Potassium 2-(1-hydroxypentyl)-benzoate (dl-PHPB) is a novel pro-drug of 3-n-butylphthalide (dl-NBP) that is used to treat ischemic stroke. Currently, dl-PHPB is in phase II–III clinical trials in China. In this study, we investigated the conversion and pharmacokinetics profiles of dl-PHPB in vitro and in vivo. The conversion of dl-PHPB to dl-NBP was pH- and calcium-dependent, and paraoxonase was identified as a major enzyme for the conversion in rat plasma. The pharmacokinetics, tissue distribution and excretion of dl-PHPB were studied and compared with equal-molar doses of dl-NBP in rats and dogs. The in vivo studies showed that dl-PHPB could be quickly and completely converted to dl-NBP. The plasma concentration-time course of converted dl-NBP after intravenous dl-PHPB administration was nearly the same as that after equal-molar dl-NBP. The Cmax and AUC of dl-NBP after oral dl-PHPB administration in rats and dogs were higher by 60% and 170%, respectively, than those after oral dl-NBP administration. Analysis of the tissue distribution of dl-PHPB revealed that converted dl-NBP was primarily distributed in fat, the brain and the stomach. In the brain, the levels of dl-NBP were relatively higher after dl-PHPB treatment by orally than after treatment with equal-molar dl-NBP. Approximately 3%–4% of dl-NBP was excreted within 72 h after dosing with dl-PHPB or dl-NBP, but no dl-PHPB was detected in urine or feces excrements. Our results demonstrate that the conversion of dl-PHPB is fast after oral or intravenous administration. Furthermore, the bioavailability of dl-PHPB was obviously better than that of dl-NBP.
Similar content being viewed by others

Introduction
Potassium 2-(1-hydroxypentyl)-benzoate (dl-PHPB), a potential pro-drug of 3-n-butylphthalide (dl-NBP) with a novel molecular structure, has been developed as an anti-cerebral ischemia agent with extremely high solubility in water1. The phase I clinical trial of dl-PHPB has been completed, and dl-PHPB is now in phase II-III clinical studies.
Dl-NBP is an oily compound with a boiling point of 140–141 °C (320 Pa) that was originally isolated from the seeds of Apium graveolensLinn, or Chinese celery2. It has been widely used for ischemic stroke and has shown good therapeutic effects in China3,4,5,6,7. However, dl-NBP is hydrophobic and is difficult to be intravenously administered, which limits the use of dl-NBP in patients with cerebral ischemic stroke7. Additionally, the bioavailability of dl-NBP is very low7. Dl-PHPB has been designed and synthesized by the Department of Medicinal Chemistry, Institute of Materia Medica, Chinese Academy of Medical Sciences as a pro-drug of dl-NBP with very good solubility in water. Dl-PHPB can be converted to an active metabolite (dl-NBP) under certain chemical (ie, acidic) and biological conditions. Pharmacological studies have demonstrated that dl-PHPB exhibited similar or stronger neuroprotective effects than dl-NBP at equal-molar doses1,8,9,10. The mechanisms underlying the anti-stroke properties of dl-PHPB include increased regional cerebral blood flow in ischemic zones and inhibited platelet aggregation1,8,9. Recent studies have shown that dl-PHPB improved cognitive defects by attenuating amyloid and tau protein pathologies in APP/PS1 transgenic mice, promoting long-term potentiation (LTP) in Aβ1-42-injected rats and APP/PS1 mice10,11,12. Additionally, dl-PHPB protected neurons against H2O2-induced apoptosis in human neuroblastoma SK-N-SH cells by modulating apoptosis-related proteins; the PKC signaling pathway may also be involved in these mechanisms13.
Although the pharmacological properties of dl-PHPB have been intensively investigated, its conversion, absorption, distribution, metabolism and excretion have not been well understood. Only a few studies have investigated the conversion of dl-PHPB in rat plasma1. Thus, the elucidation of the conversion and metabolism of dl-PHPB for clinical pharmacology and safety evaluations is very important.
In this study, we assessed in vitro studies to reveal the conversion mechanisms of dl-PHPB to dl-NBP and considered non-enzymatic acidic and alkaline solutions, calcium-dependent effect and plasma enzyme-mediated conversions. We also carried out in vivo studies to compare the pharmacokinetics, distribution and excretion of dl-PHPB with its metabolite dl-NBP after oral and intravenous administration in SD rats and Beagle dogs.
Materials and methods
Animals
SD rats (male and female, 190–210 g) were purchased from Vital River Lab Animal Technology Co, Ltd (Beijing, China), and Beagle dogs were obtained from Marshall Biotechnology Co, Ltd (Beijing, China). Animals were allowed free access to food and water in a temperature-controlled environment at 22 °C–25 °C during the experimental period. The experiments were performed in accordance with the guidelines for the care and use of laboratory animals and were approved by the Animal Care Committee of the Peking Union Medical College and the Chinese Academy of Medical Sciences (Beijing, China).
Chemicals and materials
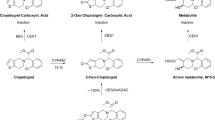
Dl-PHPB and dl-NBP were offered by the Department of Synthetic Pharmaceutical Chemistry of the Institute of Materia Medica (Beijing, China) with purities of 99.1%, and 99.0%, respectively. The chemical structures of the two compounds are shown in Figure 1. Internal standards of 4-biphenylacetic acid and diazepam were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (NICPBP, Beijing, China). Sodium bis-p-nitrophenyl phosphate (BNPP), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), phenylmethylsulfonyl fluoride (PMSF), teicoplanin, donepezil, ethylenediamine tetraacetic acid disodium salt dehydrate (EDTA-Na2), ethyleneglycol-bis (2-aminoethylether)-tetraacetic acid (EGTA), and sodium fluoride (NaF) were purchased from Sigma-Aldrich (St Louis, MO, USA). High-performance liquid chromatography (HPLC) grade methanol was purchased from Burdick & Jackson Company (Muskegon, MI, USA). All other reagents were of analytical grade. Deionized water used throughout the study was purified using a Millipore water purification system (Milford, MA, USA).

Structures of dl-NBP and dl-PHPB.
Fresh whole blood was collected and pooled from healthy SD rats and beagle dogs with heparin or EDTA-Na2 as an anticoagulant. Plasma was separated by centrifugation at 1123×g at 4 °C for 10 min. All pooled plasma was stored at −70 °C and thawed at 4 °C before assays.
Conversion of dl-PHPB in solutions of different pH values
Appropriate amounts of deionized water, 1.0 mol/L HCl, and 1.0 mol/L NaOH were used to make solutions with 1.0, 3.0, 7.0, and 9.0 pH. Reactions were initiated by adding an aliquot of a stock solution of dl-PHPB (1 mg/mL, in methanol) to obtain a final concentration of 10 μg/mL. The samples were allowed to settle for various time intervals from 0 to 360 min at 37 °C. An internal standard at a final concentration of 5 μg/mL was added to each sample, and the samples were analyzed without any pre-treatment by HPLC as described below. A control sample of dl-PHPB (10 μg/mL in methanol) or dl-NBP (7.8 μg/mL in methanol) was prepared and analyzed. For each sample, three replicated operations were performed. Residual percentage versus pH value was plotted, and degradation trends were observed.
Residual% of dl-PHPB or dl-NBP was calculated as follows:
Residual%=A/B (control)×100%
where A represents the concentration of dl-PHPB or dl-NBP produced by the solutions with different pH values and various time intervals, and B represents the control concentration of dl-PHPB or dl-NBP.
Conversion of dl-PHPB in SD rat plasma
SD rat plasma with heparin as an anticoagulant was pre-incubated at 37 °C for 5 min. The reaction was initiated by adding dl-PHPB solution (1 mg/mL, in methanol) to achieve a final concentration of 10 μg/mL. The temperature was maintained at 37 °C, representing body temperature, for the treatment of the plasma samples. A control sample of dl-PHPB (10 μg/mL in methanol) or of dl-NBP (7.8 μg/mL in methanol) was prepared. At different time intervals (0 to 360 min) at 37 °C, a sample of 200 μL was withdrawn. The reaction was stopped by adding 50 μL of saturated EDTA-Na2 solution (0.3 mol/L) and by placing the samples on ice. For each sample, three replicated operations were performed. Residual percentage versus time was plotted. The quantifications of dl-PHPB and its active metabolite (dl-NBP) in these samples were performed by HPLC methods as described below.
Residual % of dl-PHPB or dl-NBP was calculated as follows:
Residual%=A/B (control)×100%
where A represents the concentration of dl-PHPB or dl-NBP produced by the incubation in rat plasma with different time intervals, and B represents the control concentration of dl-PHPB or dl-NBP.
Effects of esterase inhibitors on dl-PHPB conversion
To characterize the esterases responsible for dl-PHPB conversion, dl-PHPB was incubated in rat plasma with heparin as an anticoagulant in the presence of various esterase inhibitors. This assay was conducted according to described methods, with modifications14,15,16,17. Briefly, the rat plasma (100 μL) was mixed with 350 μL of 50 mmol/L Tris-HCl buffer (pH 7.4) and either 50 μL of the inhibitor solution (in 10% v/v DMSO in water) or 50 μL of 10% DMSO (as control). The reaction was initiated by adding 20 μL of dl-PHPB solution (300 μg/mL, in methanol). The mixtures were incubated at 37 °C for 2 h. The reaction was stopped using 80 μL of saturated EDTA-Na2 solution, and the tubes were placed on ice.
The quantification of dl-PHPB and dl-NBP in these samples was performed by HPLC methods as described below. The conversion of dl-PHPB was calculated as follows:
Conversion of dl-PHPB%=A/B (control)×100%
where A and B (control) represent the dl-NBP amounts produced by the incubation with and without the inhibitor, respectively.
Conversion of dl-PHPB in SD rats
The vivo conversion of dl-PHPB was studied in SD rats with intravenous injections of 20 mg/kg dl-PHPB. The plasma concentration time curves of dl-PHPB and its metabolite dl-NBP were measured by HPLC methods.
Briefly, 300 μL blood samples from the femoral vein were collected into tubes containing 10 μL of saturated EDTA-Na2 at different time intervals after dosing. The blood samples were immediately centrifuged at 1123×g for 5 min, and the plasma was stored at −70 °C until HPLC analysis.
Plasma pharmacokinetics
Comparative studies on plasma pharmacokinetics of dl-PHPB and dl-NBP were carried out in Beagle dogs and SD rats. According to previous reports1, the effective doses of dl-PHPB in rats subjected to acute focal cerebral ischemia-reperfusion were 5–10 mg/kg and 50–100 mg/kg by intravenous or oral administration, respectively. Thus, the following dosage ranges were selected to cover the effective dose of dl-PHPB. The concentration time curves of dl-PHPB and its active metabolite (dl-NBP) were measured after treatment with dl-PHPB intravenously (5, 10, and 20 mg/kg in SD rats or 2, 4, and 8 mg/kg in Beagle dogs) and orally (50, 100, and 200 mg/kg in SD rats or 10, 30, and 100 mg/kg in Beagle dogs). According to the molecular weights of the two compounds, dl-NBP was given at 15.6 mg/kg in SD rats and 6.2 mg/kg in Beagle dogs intravenously and at 156.0 mg/kg in SD rats and 23.4 mg/kg in Beagle dogs orally. Then, 300 μL blood samples were collected at different time intervals after dosing, according to the procedure described above. The plasma was stored at −70 °C until HPLC analysis.
Tissue distribution and excretion
SD rats were given dl-PHPB (10 mg/kg intravenously or 100 mg/kg orally) or equimolar doses of dl-NBP (7.8 mg/kg intravenously or 78 mg/kg orally). Tissues from the heart, liver, spleen, lung, kidney, stomach, small intestine, fat, and brain were obtained at 20, 60, 120, and 240 min by oral administration and at 20, 60, and 240 min by intravenous administration, respectively. Urine and feces samples were collected pre-dose and at 0–4, 4–8, 8–12, 12–24, and 24–36 h or at 24–48, 36–48, and 48–72 h post-dose. The total urine volume and total feces weight were recorded after each sample collection. The tissue, urine and feces samples were stored at −70 °C until analysis.
Quantitative analysis of dl-PHPB and dl-NBP
Preparation of plasma and urine samples
For every 100 μL aliquot of pooled plasma (or urine) samples, 300 μL of methanol containing 10 μg/mL of internal standard were added. Mixtures were mixed for 1 min and centrifuged for 10 min at 12 000×g. A 200 μL aliquot of the supernatant was taken and filtered through a 0.22-μm membrane. A 60 μL aliquot was used for HPLC analysis.
Preparation of tissues and feces samples
Each feces sample was thawed and placed in 5 parts (1 g: 5 mL) methanol. The mixture was homogenized by a motor-driven homogenizer (Fisher Scientific, Pittsburgh, PA, USA) and subjected to ultrasound treatment for 15 min. A 5 mL aliquot of the mixture was removed and centrifuged at 3500×g for 10 min. A 400 μL aliquot of the supernatant was taken and filtered through a 0.22-μm membrane. A 60 μL aliquot was used for HPLC analysis.
HPLC analysis
The concentrations of dl-PHPB and dl-NBP in plasma were determined by previously described HPLC methods18,19,20. An Agilent 1100 (Palo Alto, CA, USA) instrument was used. Standard curves with concentrations ranging from 0.05 to 60 μg/mL dl-PHPB and 0.01 to 60 μg/mL dl-NBP in plasma and urine exhibited good linearity with correlation coefficients >0.998. The limits of the dl-PHPB and dl-NBP assays were 0.05 μg/mL and 0.01 μg/mL, respectively, with acceptable precision and accuracy. The assay precision (deviation) was <15%, whereas the assay accuracy was 85%–115%. The mean absolute percentage recoveries of dl-PHPB and dl-NBP were more than 70% from spiked plasma, urine, tissues, and feces samples determined at different concentrations of quality control (QC) samples. QC samples with three concentration levels were prepared in control stabilized samples at final concentrations of 0.5, 10, and 50 μg/mL for dl-PHPB and 0.1, 10, and 50 μg/mL for dl-NBP. Intra- and inter-day plasma assay variability were less than 10%. HPLC methods for the determinations of dl-PHPB or dl-NBP were reliable and reproducible because the % CV and % bias were below 15% for all theoretical concentrations of dl-PHPB and dl-NBP.
Data analysis
First order rate constants (K) of conversion were calculated from the slopes of linear plots of residue percentage against time, and the corresponding half-life was obtained from the equation: t1/2=0.693/K. All values were obtained from three independent determinations, and the mean values of the constants were calculated. The pharmacokinetic parameters were analyzed using 3P97 computer software package (Chinese Pharmacology Society, Beijing, China). The statistical analysis of outcome parameters was performed using SPSS version 13.5 (SPSS Inc, Chicago, IL, USA). The statistical evaluation was performed using analysis of variance (ANOVA) followed by multiple comparison tests by Duncan's method. The level of significance was set at P<0.05.
Results
Conversion of dl-PHPB to dl-NBP in vitro and in vivo
The in vitro and in vivo conversions of dl-PHPB to dl-NBP are shown in Figure 2. To study the in vitro conversion, dl-PHPB was added to distilled water of different pH values (1.0, 3.0, 7.0, and 9.0) or rat plasma and incubated at 37 °C for different periods. The in vitro conversion of dl-PHPB into dl-NBP showed a significant pH dependency. The level of dl-PHPB decreased rapidly and was undetectable at pH 1.0 after 30 min of incubation, whereas the level of dl-NBP increased. However, dl-PHPB was stable in neutral and alkaline solutions. The residue concentrations of dl-PHPB were 93% and 96% at initial pH levels of 7.0 and 9.0 after 4 h of incubation, respectively. At the same time, lower levels of dl-NBP (approximately 7% and 5% of the maximum) were detected in the solutions (Figures 2A, 2B). The conversion rates agreed with the first order kinetics shown in Table 1. The conversion in rat plasma was even more rapid. After 20 min of incubation in rat plasma at 37 °C, more than 90% of dl-PHPB was converted into dl-NBP (Figure 2C).

Time profile of dl-PHPB conversion. Time profiles of dl-PHPB and dl-NBP concentration in different pH solutions (A: dl-PHPB, B: dl-NBP) and SD rat plasma (C) were obtained after incubation at a final concentration of 10 μg/mL for different times at 37 °C (each point represents the mean±standard error of triplicate determinations). In vivo conversion (D), the mean plasma concentration versus time profiles of dl-PHPB and dl-NBP were obtained after the intravenous administration of dl-PHPB (20 mg/kg) in SD rats (n=6, 3/sex). Each point represents the mean±SD.
The in vivo conversion studies were carried out in SD rats. Dl-PHPB was intravenously injected at 20 mg/kg body weight. The mean plasma concentration time curves of dl-PHPB and its active metabolite are shown in Figure 2D. After injecting dl-PHPB, the conversion of dl-PHPB to dl-NBP was very rapid in the rats and could not be detected after 30 min; dl-NBP appeared at high levels immediately after injection. The concentrations of the active metabolite dl-NBP were at least 3-fold higher than those of dl-PHPB at the same time points. The pharmacokinetic parameters of the active metabolite dl-NBP are listed in Table 2. A two-compartment open model provided the best fit to the plasma concentration time profiles of dl-NBP obtained in rats after intravenous treatment with dl-PHPB. For this kinetic model, t1/2α and t1/2β were 4.33 and 44.76 min, respectively.
Conversion of dl-PHPB was affected by esterase inhibitors
Due to the rapid conversion of dl-PHPB in rat plasma, the esterases mediating dl-PHPB metabolism were investigated by using specific inhibitors. As shown in Figure 3, in rat plasma, a nonspecific esterase inhibitor (NaF, 200 mmol/L) and metal ion chelating agents (EDTA-Na2, 10 mmol/L and EGTA, 10 mmol/L) were highly effective inhibitors of dl-PHPB conversion (>90%). DTNB (arylesterase inhibitor, 1 mmol/L), PMSF (serine esterase inhibitor, 1 mmol/L) and BNPP (carboxylesterase inhibitor, 1 mmol/L) showed little or no suppression of the conversion of dl-PHPB. Interestingly, PMSF did not inhibit the degradation of dl-PHPB but was a relatively effective inhibitor of cholinesterase21. Donepezil (another cholinesterase inhibitor, CHEI) and teicoplanin (paraoxonase inhibitor, PONI) showed significant inhibition in concentration-dependent manners (P<0.05). These results indicated that paraoxonase was likely responsible for the lactonization of dl-PHPB to form its active metabolite dl-NBP in blood. This result was consistent with the inhibitory effects of NaF in rat plasma; 200 mmol/L of NaF was sufficient to inhibit PON activity22.

Effects of various esterase inhibitors on dl-PHPB conversion in rat plasma. The conversion of dl-PHPB to dl-NBP was highly effectively inhibited by NaF (200 mmol/L, a general esterase inhibitor) (A), and EDTA or EGTA (10 mmol/L, metal ion chelating agents) (B); partly inhibited by teicoplanin (1 mmol/L, a paraoxonase inhibitor) (C), donepezil (1 mmol/L, a cholinesterase inhibitor) (D); and negligibly inhibited by DTNB (1 mmol/L, an arylesterase inhibitor) (E), BNPP (1 mmol/L, a carboxylesterase inhibitor) (F) or PMSF (1 mmol/L, a serine esterase inhibitor) (G) (values are the mean±standard error of triplicate determinations). *P<0.05, **P<0.01 compared with control group.
Comparative plasma pharmacokinetics
The comparative pharmacokinetic studies of dl-PHPB and dl-NBP were carried out at equal-molar doses. The plasma concentration-time profiles of dl-NBP were determined following intravenous or oral administration of dl-PHPB and dl-NBP in SD rats and Beagle dogs. Figures 4A and 4B show that the concentration time curves of dl-NBP, whether converted from dosing dl-PHPB or from direct intravenous administration of dl-NBP, were nearly identical in dogs and in rats, implying a rapid transformation of dl-PHPB to dl-NBP in blood. The main pharmacokinetic parameters of dl-NBP after intravenous administration of dl-PHPB or dl-NBP are summarized in Table 2. The parameters were very similar at equal-molar doses, and the areas under the curve (AUCs) were nearly the same. However, after the oral administration of dl-PHPB, the peak concentrations and the AUCs of dl-NBP in both animal models were significantly higher than those after the oral administration of dl-NBP. This indicated that dl-PHPB was quickly absorbed and the AUC was higher than that of dl-NBP (Figures 4C and 4D). The main pharmacokinetic parameters of dl-NBP are listed in Table 3. At equal-molar doses, ie, 30 mg/kg of dl-PHPB and 23.4 mg/kg of dl-NBP in dogs and 200 mg/kg of dl-PHPB and 156 mg/kg of dl-NBP in rats, the time to peak (tpeak) was faster and the Cmax and AUC were higher in the dl-PHPB groups (greater by approximately two-fold or higher) than those in the dl-NBP groups. Other parameters did not show any significant differences.

Comparative plasma concentration versus time profiles of dl-NBP after administration of dl-PHPB and dl-NBP. Based on the molecular weights of two compounds, dl-PHPB and dl-NBP were intravenously administered to SD rats (A: 20 mg/kg for dl-PHPB, and 15.6 mg/kg for dl-NBP) or Beagle dogs (B: 8 mg/kg for dl-PHPB, and 6.2 mg/kg for dl-NBP); and orally administered in SD rats (C: 200 mg/kg for dl-PHPB, and 156 mg/kg for dl-NBP) and Beagle dogs (D: 30 mg/kg for dl-PHPB, and 23.4 mg/kg for dl-NBP). Blood samples were obtained at 0, 1, 3, 5, 10, 15, 20, 30, 45, 60, 120, and 240 min after administration. Values are mean±standard deviation (n=6, 3/sex).
Dose-dependent pharmacokinetics were also studied. The plasma concentration time curves of dl-NBP in both animal models after the intravenous or oral administration of dl-PHPB are shown in Figure 5, and the pharmacokinetic parameters are provided in Tables 2 and 3. The data present a linear exposure value in the metabolism of dl-NBP in beagle dogs and a dose proportional exposure value in SD rats after intravenous or oral administration of dl-PHPB. We also tried to detect the plasma concentration time profiles of dl-PHPB. However, the conversion of dl-PHPB in plasma was too rapid, and the concentration was too low to be detected.

Mean plasma concentration versus time profiles of dl-NBP after the administration of dl-PHPB. Dl-PHPB was intravenously administered in Beagle dogs (A: 2, 4, and 8 mg/kg) and SD rats (B: 5, 10, and 20 mg/kg); and orally administered in Beagle dogs (C: 10, 30, and 100 mg/kg) and SD rats (D: 50, 100, and 200 mg/kg). Blood samples were obtained at 0, 1, 3, 5, 10, 15, 20, 30, 45, 60, 120, and 240 min after administration. Values are the mean±standard deviation (n=6, 3/sex).
Distribution in tissues
Figure 6 shows the distributions of dl-NBP in rat tissues after intravenous or oral treatment with dl-PHPB or dl-NBP. The levels of dl-NBP were determined in heart, liver, lung, spleen, kidney, brain, stomach, fat, and small intestine tissues at different times after treatment. The results showed that the parent drug dl-NBP was primarily distributed in the stomach, kidney, fat, and brain tissues after either oral or intravenous treatment with dl-PHPB or dl-NBP. The highest concentrations of dl-NBP appeared in the stomach and in fat after oral or intravenous treatment. In brain distribution, relatively higher levels were observed at different times after dl-PHPB treatment than after dl-NBP treatment, especially with oral administration; maximum concentrations of 40.6±8.9 μg/g at 60 min and 25.4±11.9 μg/g were observed, respectively.

Tissue distribution of dl-NBP after equimolar dosing of dl-PHPB and dl-NBP in SD rats. The tissues were obtained at 20, 60, and 240 min after dl-PHPB (A: 10 mg/kg) or dl-NBP (B: 7.8 mg/kg) injection or at 20, 60, 120, and 240 min after oral dl-PHPB (C: 100 mg/kg) or dl-NBP (D: 78 mg/kg) administration. Values are the mean±standard deviation (n=6, 3/sex).
Excretion of dl-PHPB
After equal-molar doses of dl-PHPB or dl-NBP by oral or intravenous administration, the cumulative recovery of dl-NBP in urine and feces of SD rats is shown in Tables 4 and 5. Less than 5% of dl-NBP was recovered in urine and feces after oral treatment of either compound within 72 h. Dl-PHPB was undetectable in the urine and feces of the dl-PHPB group. The excretion rates of dl-NBP in the urine or feces were similar between the two compounds, by either oral or intravenous administration, which implied that dl-PHPB was converted to dl-NBP and was then excreted as metabolites.
Discussion
The elucidation of the conversion mechanism of pro-drugs in chemical and biological samples is essential for enhancing pharmacological efficacy and safety. Dl-PHPB is liable to lactonization of the hydroxyl acid to form the pharmacologically active metabolite dl-NBP. The oral AUC value of the converted dl-NBP from dl-PHPB was two- to three-fold greater than that of dl-NBP directly administered.
Esters are typically unstable compounds in vivo due to esterases in bio-matrices23. Studies have identified in vitro hydrolyzing ester compounds in bio-samples that are involved in the in vivo metabolic pathway24,25,26,27,28. Non-enzymatic conversions indicate that chemical lactonization can occur. This pH-dependent conversion is important due to the various pH conditions encountered in the stomach (pH 2.0), bile (pH 8.5), plasma (pH 7.4) and intestines (pH 6.8). Because dl-PHPB is quite stable in alkaline conditions, dl-PHPB could be stored (could not be converted) in alkaline aqueous solutions.
The conversion rate was more rapid in rat plasma samples than in non-enzymatic solutions, indicating that enzymatic lactonization is the key mode of conversion in plasma. The conversion was markedly inhibited in rat plasma by NaF, a general esterase inhibitor14,22,29, confirming that one or several plasma esterases are involved in dl-PHPB metabolism. Moreover, the conversion in rat plasma was almost completely inhibited by EDTA and EGTA (a general chelating agent for calcium) at 10 mmol/L and significantly restrained by donepezil (AChE inhibitor)30 and teicoplanin (PON inhibitor) in a concentration-dependent manner31. However, PMSF, another inhibitor of AChE21, did not show any inhibitory effects on dl-PHPB transformation. Additionally, due to the presence of calcium-dependent lactonase in sera23,32, EDTA and EGTA are potent inhibitors of PON and arylesterase via calcium ion chelation, which is essential for their hydrolytic activity33,34. Thus, the results suggested that PON may be primarily responsible for the conversion of dl-PHPB in the plasma esterase.
The HPLC analyses of plasma samples after intravenous or oral treatment indicated a rapid and complete conversion of dl-PHPB to its active form (dl-NBP) in blood, which was consistent with the conversion in vitro studies (in non-enzymatic acidic solution or rat plasma). As described above, the conversion of dl-PHPB in plasma was too fast to be detected. After intravenous or oral dosing, the concentrations of dl-PHPB in plasma rapidly declined within 30 min, whereas dl-NBP concentrations remained at higher levels until 240 min. Thus, the metabolism of dl-PHPB, such as plasma pharmacokinetics, tissue distribution, and excretion, were carried out on its active form (dl-NBP) of dl-PHPB in SD rats and Beagle dogs. Compared with the oral administration of equal-molar doses of dl-NBP, the total drug exposure expressed as AUC and the Cmax of the converted dl-NBP were two-fold higher in SD rats and Beagle dogs after the oral administration of dl-PHPB. We also found that the pharmacokinetic parameters of dl-NBP following intravenous administration of dl-PHPB in rats and dogs were similar to those of dl-NBP based on equal-molar dosages. Thus, the pharmacokinetic profile of dl-PHPB showed a rapid absorption, and the converted dl-NBP showed a higher bioavailability.
The distribution study showed that the concentrations of dl-NBP were low or undetected in all tissues after 240 min, indicating no long-term accumulation after oral dl-PHPB administration. However, there was a relatively higher distribution in fat tissue than in dl-NBP-treated animals after intravenous dosing of dl-PHPB. These effects may help to maintain plasma drug concentrations and may result in higher bioavailability. When dl-PHPB was orally administered, the distribution of converted dl-NBP in rat brain was enhanced by 60% compared with that observed in oral equal-molar doses of dl-NBP. The high distribution in the brain confirmed the pharmacological effects of dl-NBP on anti-cerebral ischemia1,5,6,7 and the therapeutic benefit of dl-PHPB.
Our study showed that no original form of dl-PHPB was excreted and only less than 5% of dl-NBP was recovered in the urine and feces after the administration of dl-PHPB in SD rats within 72 h. Because of the rapid and complete conversion of dl-PHPB to dl-NBP, this may indicate that the converted dl-PHPB to dl-NBP in blood or tissues was primarily excreted in the parent-drug form. A low percentage of the parent-drug form was detected in urine and feces. Similar observations have been reported in rats after the administration of dl-NBP35,36. Within 24 h following oral3H]-NBP administration, the total radioactivity in urine and feces was 73.1% of the original dose, and the total prototype drug excreted in urine and feces was 2.53% of the dose36. Furthermore, 23 metabolites of dl-NBP were detected in human plasma and urine37. Thus, the metabolic pathway of dl-PHPB in vivo may be similar to that of dl-NBP. Additionally, dl-PHPB is a weakly basic drug, exhibits pH-dependent stabilization and is susceptible to gastric and intestinal (GI) tract pH change. The varying GI pH levels may also contribute to differences in drug absorption. The rate of gastric acid secretion is known to be widely different among species. For example, rats and humans have good acid secretions but dogs have poor secretions38,39,40,41. In the present study, the results showed that the bioavailability of dl-PHPB in dogs was higher than that in rats. The differences in bioavailability between dogs and rats are most likely due to low basal gastric acid secretions increasing GI pH and enhancing the stabilization of dl-PHPB in this species. Further studies are required to clarify the effects of pH changes on the metabolism of dl-PHPB, conversion mechanisms, metabolic pathways for dl-PHPB, and potential interactions with other drugs.
In conclusion, as a pro-drug, dl-PHPB was rapidly and completely converted into dl-NBP after treatment in rats and dogs. The Cmax and AUC values of dl-NBP converted from dl-PHPB were significantly higher than those from direct dl-NBP administration. Conversion in vivo was primarily mediated by plasma esterases. The active form was more distributed in fat and brain tissues. The dl-PHPB metabolic pathway after conversion may follow that of dl-NBP.
Author contribution
Xiao-liang WANG conceived and designed the experiments; Jiang LI, Shao-feng XU, Nan FENG, and Ling WANG performed the experiments; Jiang LI wrote the paper. Xiao-liang WANG, Jiang LI, and Ying PENG analyzed the data.
References
Zhang Y, Wang L, Li J, Wang XL . 2-(1-Hydroxypentyl)-benzoate increases cerebral blood flow and reduces infarct volume in rats model of transient focal cerebral ischemia. J Pharmacol Exp Ther 2006; 317: 973–9.
XU Z, HU GY, TAN GS . Comparative research on methods of n-butylphthalide synthesis. China J Modern Med 2004; 14: 91–3.
Wang XW . 3-n-Butylphthalide. Drugs Future 2000; 25: 16–23.
Dong GX, Feng YP . Effects of NBP on ATPase and anti-oxidant enzymes activities and lipid peroxidation in transient focal cerebral ischemic rats. Acta Acad Med Sin 2002; 24: 93–7.
Cui LY, Liu XQ, Zhu YC, Fan DS, Xie RP, Shen Y, et al. Effects of dl-3-butylphthalide on treatment of acute ischemic stroke with moderate symptoms: a multi-center, randomized, double-bind, placebo-control trial. Chin J Neurol 2005; 38: 251–4.
Cerebrovascular Disease Working Groups, Chinese Academy of Neurology. Guidelines for the management of iscemic stroke (China 2010). Chin J Neurol 2010; 43: 146–53.
Xu B, Zhao ZG . Butylphthalide injection, an innovative drug originated in China for the treatment of ischemic stroke. Chin J New Drugs 2011; 20: 947–50.
Zhang Y, Wang L, Zhang LY, Wang XL . Effects of 2-(1-hydroxypentyl)-benzoate on platelet aggregation and thrombus formation in rats. Drug Dev Res 2004; 63: 174–80.
Yang HY, Xu SF, Li J, Wang L, Wang XL . Potassium 2-(1-hydroxypentyl)-benzoate inhibits ADP-induced rat platelet aggregation through P2Y1-PLC signaling pathways. Naunyn Schmiedebergs Arch Pharmacol 2015; 388: 983–90.
Zhao WH, Xu SF, Peng Y, Ji XC, Cao DX, Li J, et al. Potassium 2-(1-hydroxypentyl)-benzoate improves learning and memory deficits in chronic cerebral hypoperfused rats. Neurosci Lett 2013; 541: 155–60.
Li PP, Wang WP, Liu ZH, Xu SF, Lu WW, Wang L, et al. Potassium 2-(1-hydroxypentyl)-benzoate promotes long-term potentiation in Aβ1-42-injected rats and APP/PS1 transgenic mice. Acta Pharmacol Sin 2014; 35: 869–78.
Peng Y, Hu YL, Xu SF, Rong XF, Li J, Li PP, et al. Potassium 2-(1-hydroxypentyl)-benzoate improves memory deficits and attenuates amyloid and τ pathologies in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther 2014; 350: 361–74.
Hu YL, Peng Y, Long Y, Xu SF, Feng N, Wang L, et al. Potassium 2-(1-hydroxypentyl)-benzoate attenuated hydrogen peroxide-induced apoptosis in neuroblastoma SK-N-SH cells. Eur J Pharmacol 2012; 680: 49–54.
Tsujikawa K, Kuwayama K, Miyaguchi H, Kanamori T, Iwata YT, Inoue H . In vitro stability and metabolism of salvinorin A in rat plasma. Xenobiotica 2009; 39: 391–8.
Koitka M, Höchel J, Gieschen H, Borchert HH . Improving the ex vivo stability of drug ester compounds in rat and dog serum: Inhibition of the specific esterases and implications on their identity. J Pharm Biomed 2010; 51: 664–78.
Gil F, Gonzalvo MC, Hernandez AF, Villanueva E, Pla A . Differences in the kinetic properties, effect of calcium and sensitivity to inhibitors of paraoxon hydrolase activity in rat plasma and microsomal fraction from rat liver. Biochem Pharmacol 1994; 48: 1559–68.
Sogorb MA, Sánchez I, López-Rivadulla M, Céspedes V, Vilanova E . EDTA-resistant and sensitive phosphotriesterase activities associated with albumin and lipoproteins in rabbit serum. Drug Metab Dispos 1999; 1: 53–9.
Li J, Wang XL, Wang AP, Xu SF, Jin HT . Toxicokinetics and toxicity of potassium 2-(1-hydroxypentyl)-benzoate in beagle dogs. J Asian Nat Prod Res 2017; 19: 388–401.
Li J, Wang AP, Wang XL, Xu SF, Li J, Li H, et al. Experimental study on repeated dose toxicity and toxicokinetics of dl-PHPB by intravenous injection in Beagle dogs. Modern Med J China 2011; 13: 1–3.
Li J, Wang AP, Wang XL, Xu SF, Li J, Li H, et al. Experimental study on repeated dose toxicity and toxicokinetics of dl-PHPB by intravenous injection in Beagle dogs. Modern Med J China 2011; 13: 1–3.
Turini P, Kurooka S, Steer M, Corbascio AN, Singer TP . The action of phenylmethylsulfonyl fluoride on human acetylcholinesterase, chymotyrpsin and trypsin. J Pharmacol Exp Ther 1969; 167: 98–104.
Dean RA, Christian CD, Sample RHB, Bosron WF . Human liver cocaine esterases: ethanol-mediated formation of ethylcocaine. FASEB J 1991; 5: 2735–9.
Liederer BM, Borchardt RT . Enzymes involved in the bioconversion of ester-based prodrugs. J Pharm Sci 2006; 95: 1177–95.
Williams FM, Moore U, Seymour RA, Mutch EM, Nicholson E, Wright P, et al. Benorylate hydrolysis by human plasma and human liver. Br J Clin Pharmacol 1989; 28: 703–8.
Li W, Escarpe PA, Eisenberg EJ, Cundy KC, Sweet C, Jakema KJ, et al. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob Agents Ch 1998; 42: 647–53.
Prueksaritanont T, DeLuna P, Gorham LM, Ma B, Cohn D, Pang J, et al. In vitro and in vivo evaluations of intestinal barriers for the zwitterion L-767,679 and its carboxyl ester prodrug L-775,318. Roles of efflux and metabolism. Drug Metab Dispos 1998; 26: 520–7.
Ericsson H, Tholander B, Regårdh CG . In vitro hydrolysis rate and protein binding of clevidipine, a new ultrashort-acting calcium antagonist metabolised by esterases, in different animal species and man. Eur J Pharm Sci 1999; 8: 29–37.
Anand SS, Bruckner JV, Haines WT, Muralidhara S, Fisher JW, Padilla S . Characterization of deltamethrin metabolism by rat plasma and liver microsomes. Toxicol Appl Pharm 2006; 212: 156–66.
Wei RD, Chu FS . Modification of in vitro metabolism of T-2 toxin by esterase inhibitors. Appl Environ Microb 1985; 50: 115–9.
Bryson HM, Benfield P . Donepezil. Drugs Aging 1997; 10: 234–49, discussion 240-1.
Ekinci D, Beydemir S . Evaluation of the impacts of antibiotic drugs on PON 1; a major bioscavenger against cardiovascular diseases. Eur J Pharmacol 2009; 617: 84–9.
Roth RH, Levy R, Giarman NJ . Dependence of rat serum lactonase upon calcium. Biochem Pharmacol 1967; 16: 596–8.
Gonzalvo MC, Gil F, Hernández AF, Villanueva E, Pla A . Inhibition of paraoxonase activity in human liver microsomes by exposure to EDTA, metals and mercurials. Chem-Biol Interact 1997; 105: 169–79.
Kuo CL, La Du BN . Calcium binding by human and rabbit serum paraoxonases. Structural stability and enzymatic activity. Drug Metab Dispos 1998; 26: 653–60.
Peng SH, Zhou TH . Investigation on in vivo metabolism of 3-n-butylphthalide. Acta Pharm Sin 1996; 31: 780–4.
Wang CH, Feng YP, Wu YL . A study on the metabolites of dl-3-N-butyphthalide in rats. Acta Pharm Sin 1997; 32: 641–6.
Diao XX, Deng P, Xie C, Li XL, Zhong DF, Zhang YF . Metabolism and pharmacokinetics of 3-n-butylphthalide (NBP) in humans: the role of cytochrome P450s and alcohol dehydrogenase in biotransformation. Drug Metab Dispos 2013; 41: 430–44.
Pappas TN, Debas HT, Goto Y, Taylor IL . Peptide YY inhibits meal-stimulated pancreatic and gastric section. Am J Physiol 1985; 248: G118–23.
Emås S, Grossman MI . Response of Heidenhain pouch to histamine, gastrin and feeding before and after truncal vagotomy in dogs. Scand J Gastroenterol 1969; 4: 497–503.
Petersen H, Grossman MI . Stimulation of gastric acid secretion by dimaprit in unanesthetized rats. Agents Actions 1978; 8: 566–7.
Wormsley KG, Grossman ME . Maximal histalog test in control subjects and patients with peptic ulcer. Gut 1965; 6: 427–35.
Acknowledgements
This study was supported by grants from the National Science and Technology Major Special Project on Major New Drug Innovation of China (No 2012ZX09301002-004) and the Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (No BZ0150).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, J., Xu, Sf., Peng, Y. et al. Conversion and pharmacokinetics profiles of a novel pro-drug of 3-n-butylphthalide, potassium 2-(1-hydroxypentyl)-benzoate, in rats and dogs. Acta Pharmacol Sin 39, 275–285 (2018). https://doi.org/10.1038/aps.2017.90
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.90
