
Abstract
Xyloketal B (Xyl-B) is a novel marine compound isolated from mangrove fungus Xylaria sp. We previously demonstrated that pretreatment with Xyl-B exerted neuroprotective effects and attenuated hypoxic-ischemic brain injury in neonatal mice. In the present study we investigated the neuroprotective effects of pre- and post-treatment with Xyl-B in adult mice using a transient middle cerebral artery occlusion (tMCAO) model, and explored the underlying mechanisms. Adult male C57 mice were subjected to tMCAO surgery. For the pre-treatment, Xyl-B was given via multiple injections (12.5, 25, and 50 mg·kg−1·d−1, ip) 48 h, 24 h and 30 min before ischemia. For the post-treatment, a single dose of Xyl-B (50 mg/kg, ip) was injected at 0, 1 or 2 h after the onset of ischemia. The regional cerebral perfusion was monitored using a laser-Doppler flowmeter. TTC staining was performed to determine the brain infarction volume. We found that both pre-treatment with Xyl-B (50 mg/kg) and post-treatment with Xyl-B (50 mg/kg) significantly reduced the infarct volume, but had no significant hemodynamic effects. Treatment with Xyl-B also significantly alleviated the neurological deficits in tMCAO mice. Furthermore, treatment with Xyl-B significantly attenuated ROS overproduction in brain tissues; increased the MnSOD protein levels, suppressed TLR4, NF-κB and iNOS protein levels; and downregulated the mRNA levels of proinflammatory cytokines, including IL-1β, TNF-α, IL-6 and IFN-γ. Moreover, Xyl-B also protected blood-brain barrier integrity in tMCAO mice. In conclusion, Xyl-B administered within 2 h after the onset of stroke effectively protects against focal cerebral ischemia; the underlying mechanism may be related to suppressing the ROS/TLR4/NF-κB inflammatory signaling pathway.
Similar content being viewed by others

Introduction
Stroke is one of the most common causes of death and disabilities worldwide1. The mechanisms involved in the pathophysiology of strokes are not fully understood, and thus, there is a lack of effective pharmacological therapeutic drugs. The only drug approved by the Food and Drug Administration (FDA) is tissue-plasminogen activator (tPA)2. However, tPA displays certain severe side effects and has a very limited time window, which renders it applicable only in less than 10% of all stroke patients3. Thus, there is an urgent need for new therapeutic drugs.
There is increasing evidence demonstrating that inflammation appears to play an important role in the pathogenesis of ischemic strokes and other forms of neurodegenerative brain injuries. Stroke patients who suffer from inflammation are closely associated with poorer clinical outcomes2. Experimentally, there was a time-dependent recruitment and activation of inflammatory cells in focal cerebral ischemic strokes, while inhibiting the inflammatory response decreased the infarct volume and improved the neurological deficit4. Toll-like receptors (TLRs) belong to the transmembrane pattern-recognition receptor family and play important roles in the induction and regulation of immune/inflammatory responses. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, plays a key role in ischemic strokes by activating nuclear factor kappa beta (NF-κB) and, thus, inducing the excessive generation of inflammatory factors5. The inhibition of the TLR4-NF-κB signaling pathway protects the brain from neurodegeneration in MCAO rats6. NF-κB belongs to a family of transcription factors, consisting of five subunits (p50, RelA/p65, c-Rel, RelB and p52), that form homo- or hetero-dimers. The activation of TLR4 leads to the nuclear translocation of NF-κB5 and regulates the transcription of genes related to inflammation, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6). Moreover, following cerebral ischemia, NF-κB rapidly increases in neurons and glial cells7. Hence, the downregulation of the TLR4-NF-κB signaling pathway could reduce the production of inflammatory factors and, thus, serve as a potential drug target for the treatment of strokes.
Xyloketal B (Xyl-B) is a novel marine compound that is isolated from mangrove fungus8. We first reported the protective effects of Xyl-B on an oxLDL (oxidatively modified low density lipoprotein)-induced endothelial injury9. Furthermore, Xyl-B was shown to have multiple biomedical activities, including neuroprotective effects against toxicity, antioxidant effects on endothelial cells and zebrafish, anti-glioma effects through the inhibition of TRPM7 and the reduction of atherosclerosis plaque formation in apolipoprotein E-deficiency mice10,11,12,13,14,15,16. Our previous study also demonstrated that Xyl-B protected against neonatal brain injuries induced by hypoxia-ischemia17. In this study, we further used the tMCAO adult mice model to investigate the neuroprotective effects of Xyl-B in adult strokes and explored its potential mechanisms, including alterations in the TLR4/NF-κB signaling pathway, inflammatory response and ROS generation.
Materials and methods
tMCAO (transient middle cerebral artery occlusion) mouse model
Adult male C57 mice, weighing 20-25 g, were used in this study. A transient middle cerebral artery occlusion (tMCAO) was performed. First, the mice were anesthetized with sodium pentobarbital (Merck, New Zealand, America) at 60 mg/kg (ip). During the entire tMCAO surgery, the core body temperatures of the mice were maintained at 37 °C using a heating pad (RWD Life Science, Shenzhen, China). The left common carotid artery and external and internal carotid arteries were separated, and then, a silicone-coated 6-0 suture (Doccol, USA) was gently inserted via the lumen of external and the internal carotid artery to embed into the cerebral artery for occluding the middle cerebral artery (MCA). The distance between the bifurcation of the internal and the external carotid arteries to the middle cerebral artery was 10±0.5 mm. After 2 h of occlusion, the suture was removed to allow reperfusion. Laser Doppler Flowmetry (PeriCam Psi-Zr, Sweden) was employed to verify that the occlusion was successful (reduction of at least 75% from the baseline readings). Sham-operated mice underwent the same procedure, except the suture was inserted along the internal carotid artery and then immediately withdrawn. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All animal handling and surgery procedures were in accordance with the ethical standards of the Institutional Animal Care and Use Committee at Sun Yat-sen University and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Administration of Xyl-B
Xyl-B was dissolved in 15% propylene glycol saline (used as a vehicle control) to an injection concentration of 3 mg/mL, as used in our previous study15. As shown in the schematic illustration of the experimental procedures in Figure 1, for the preventive treatments, multiple intraperitoneal injections of Xyl-B (12.5, 25 and 50 mg.kg−1.d−1) were given at 48 h, 24 h and 30 min before the onset of the tMCAO, and the first injection was given 48 h before the onset of the tMCAO. To determine the therapeutic window of Xyl-B against a stroke, a single dose of Xyl-B (50 mg/kg, ip) was administered 0 h, 1 h and 2 h after the onset of ischemia (Figure 1).

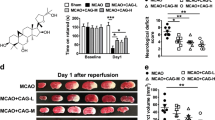
Xyl-B reduced infarct volume in the tMCAO mice. A schematic overview of the experimental procedure is shown in the upper panel. TTC staining was performed at 24 h after ischemia/reperfusion. (A) Pretreatment with multiple intraperitoneal injections of Xyl-B (12.5, 25 and 50 mg/kg) was given at 48 h, 24 h and 30 min before the onset of tMCAO. (B) A single intraperitoneal injection of Xyl-B at 50 mg/kg was given at 0 h, 1 h and 2 h after the onset of tMCAO. n=7 mice/group.
Hemodynamics of the regional cerebral perfusion
The regional cerebral perfusion was monitored using a laser-Doppler flowmeter (PeriCamPsi-Zr, Sweden). After removing the scalp, a flowmeter probe was placed on the dura and kept away from the visible cerebral vessels. The steady-state baseline values were recorded before the tMCAO. The regional cerebral perfusion was monitored continuously until a steady state was achieved after the ischemia or reperfusion. The data were normalized to the baseline values and expressed as percentages of the baseline values.
TTC staining
TTC (2, 3, 5-triphenyltetrazolium chloride, Sigma) staining was performed as described in a previous study to determine the brain infarction volume17. Briefly, the mice were rapidly sacrificed after 24 h of ischemia. The brains were isolated, frozen at -20 °C for 30 min and cut into 2.0-mm-thick slices. Coronal brain slices were stained with 2% TTC at 37°C for 10 min and then fixed in 4% paraformaldehyde for 6 h. Images of the stained slices were taken using a scanner. The infarct area in each slice was measured using the software Image-Pro Plus 6.0 (Media Cybernetics, USA). The volumes of the infarctions were calculated using the following formula: Corrected infarct volume (%)=[contralateral hemisphere volume−(ipsilateral hemisphere volume-infarct volume)]/contralateral hemisphere volume × 100%, as described in our previous study17.
Neurological deficiency assessment
The neurological status was assessed using a double-blind method according to the following 4-tiered grading system: 0 score, no observable deficits; 1 score, torso flexion to the right; 2 score, spontaneous circling to the right; 3 score, leaning/falling to the right; and 4 score, no spontaneous movement18,19.
Western blotting experiment
Western blotting was carried out as described in our previous studies9. The cytoplasmic and nuclear extracts were carried out according to the manual of the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo78833). Sample protein (50 μg) from the ipsilateral hemisphere was separated on 8% or 12% SDS-PAGE gels, which were subsequently transferred to a polyvinylidene difluoride (PVDF) membrane. Then, the membrane was immunoprobed with a TLR4 primary antibody (Santa Cruz; 1:200; sc-10741), NF-κB (p65) primary antibody (CST; 1:1000; #3031P), iNOS primary antibody (Abcam; 1:500; ab129372) and Lamin B (Santa Cruz; 1:500; sc-6216). The blots were incubated with an HRP-conjugated secondary antibody, and the signals were developed using an enhanced chemiluminescence substrate. The images of the chemiluminescence were captured with an imaging system (Bio-Rad, Hercules, and CA). The optical densities were normalized to those of β-actin, and the fold difference for each target protein was calculated as the ratio of the target protein expression/β-actin expression (ImageJ 1.42q software, U.S. National Institutes of Health).
DHE staining
The brains were cut into 8-μm-thick sections and placed on a glass slide; the tissues were processed and imaged in parallel. For this assay, the sections were dried for 30 min at 37 °C, washed 3×5 min with PBS and then incubated with DHE (0.5 μmol/L) for 30 min at room temperature in the dark. The sections were washed thrice with ice-cold PBS, and then fluorescent oxyethidium was detected (excitation/emission at 485 nm/590 nm). All sections were analyzed using a confocal system (Olympus FV500-IX81). The fluorescence intensity was normalized to the control samples without the addition of DHE.
Immunohistochemistry
For the immunohistochemistry, mice were perfused via a cardiac puncture with 4% PFA, and then, the brains were collected and sequentially cut into 8 μm thick sections . The sections were heated in a tissue-drying oven for 45 min at 60 °C, blocked with endogenous peroxidases for 15 min at room temperature, and washed 3×5 min with PBS. The sections were incubated for 30 min at 37 °C with 5% BSA. The sections were covered with the NF-κB antibody, which was diluted in the blocking buffer by 1:200, overnight at 4 °C. Then, the sections were washed 3×5 min with PBS. The sections were covered with the secondary antibody for 30 min at 37 °C and washed 3×5 min with PBS. The sections were stained with DAB for 30 s and counterstained with hematoxylin. Images were scanned using a confocal system (Olympus FV500-IX81) and analyzed by Image-Pro Plus 6.
Quantitative real-time PCR
The quantitative real-time PCR experiment was conducted as described previously20. Briefly, the total RNA from the ischemic region was isolated using the TRIzol reagent (Invitrogen). The RNA concentration was determined by a spectrophotometer (NanoDrop 2000, Thermo, Wilmington, DE, USA). In total, 1 μg of total RNA was reverse transcribed and used for the SYBR Green-based real-time RT-PCR as previously described20. The samples were run in triplicate with RNA preparations from three to five independent experiments. The QuantiTect primers are as follows: IL-1β, F/5′-AAATGCCACCTTTTGACAGTG-3′, R/5′-GAGTGATACTGCCTGCCTGA-3′; IL-6, F/5′-GACAAAGCCAGAGTCCTTCAGA-3′, R/5′-GAGCATTGGAAATTGGGGTAGG-3′; TNF-α, F/5′-TGCCTCAGCCTCTTCTCATT-3′, R/5′-GGGCTTGTCACTCGAGTTTT-3′; IFN-γ, F/5′-GTATTGCCAAGTTTGAGGTCAAC-3′, R/5′-GCTTCCTGAGGCTGGATTC-3′; and α-Tubulin, F/5′-TCTAACCCGTTGCTATCATGC-3′, R/5′-GCCATGTTCCAGGCAGTAG-3′. The real-time PCR reactions were carried out on the IQ5 Real-time PCR Detection System (Bio-Rad, USA) in a volume of 20 μL, containing 10 μL FastStart Universal SYBR green master (Roche, USA) and 0.3 μm of each primer of the tested target genes. The reaction profile consisted of 40 cycles (qPCR condition: 95°C for 15 s, 60°C for 45 s) after the initial 10-min incubation at 95°C. A dissociation stage was performed at the end of the reaction, consisting of 50 cycles of 10 s with the temperature increasing 0.5°C /cycle to demonstrate the specificity of the amplification. The mRNA expression analysis was performed in triplicate for each sample. The fold change in the mRNA expression of each gene was determined by the 2−ΔΔCt method using α-Tubulin mRNA as an internal control.
Evans blue extravasation
The Evans blue extravasation measurement was carried out according to our previous description18. Briefly, 2 h after the onset of the ischemia, 2% Evans blue (4 mL/kg, Sigma) in normal saline was injected into the tail vein of the mouse. The ipsilateral and contralateral hemispheres were separated and homogenized in 1 mL of formamide at 37 °C for 24 h. Then, the samples were centrifuged at 14000 rpm for 30 min. The Evans blue concentration in the supernatant was quantitatively determined by measuring the 630 nm absorbance.
Statistical analysis
All data are expressed as the mean±SEM. Unpaired Student's t-test was used to compare 2 groups. A one-way analysis of variance (ANOVA), followed by a Bonferroni multiple comparison post hoc test with a 95% CI, was used for 3 or more groups. The statistical significance was evaluated by the analysis of variance using Prism 4 software (GraphPad Software, San Diego, CA, USA). P<0.05 was considered statistically significant.
Results
Xyl-B reduced the infarct volume after focal ischemia
First, we determined the effect of the pretreatment with Xyl-B on brain infarction using TTC staining. As shown in Figure 1A, multiple intraperitoneal injections of different doses of Xyl-B (12.5, 25 and 50 mg/kg) were given 48 h, 24 h and 30 min before the ischemia. Pretreatment with multiple injections of Xyl-B significantly reduced the infarct volume in the tMCAO mice in a dose-dependent manner. In the sham group, the brain slices with TTC staining showed a uniform red color, indicating normal tissues. The infarct area in the brain slices from the tMCAO group showed a white color. Compared with the relative infarct volume of 26.2%±1.1% in the vehicle-treated tMCAO group (Vehicle), the relative infarct volumes in the Xyl-B pretreatment groups were 24.5%±1.6%, 20.1%±1.1% and 18.2%±1.9% at the 3 different doses of Xyl-B (12.5, 25 and 50 mg/kg, respectively),. The reduction in the infarct volume in the tMCAO groups that were pretreated with Xyl-B at 25 and 50 mg/kg/day was significantly different from that in the corresponding vehicle group (Table 1). Edaravone, which is a novel free-radical scavenger that prevents transient hypoxic-ischemic brain injury19, was used as a positive treatment control. The pretreatment with edaravone also significantly reduced the relative infarct volume in the tMCAO mice compared with that in the vehicle group.
Next, a single intraperitoneal injection of Xyl-B or vehicle at 50 mg/kg was given at 0, 1 or 2 h after the onset of the ischemia to detect its therapeutic window against a stroke. As shown in Figure 1B and summarized in Table 2, a single dose of Xyl-B at 0, 1, or 2 h after the ischemia significantly reduced the infarct volume in the tMCAO mice, and the best therapeutic effect was displayed immediately after the tMCAO insult (0 h). Accordingly, the following experiments included the administration of a single dose of 50 mg/kg Xyl-B at 0 h after the onset of the ischemia (Xyl-B treatment). As shown in Figure 2 and Table 3, after 2 h of occlusion and reperfusion, the Xyl-B treatment tended to increase the blood flow in the infarct area but did not reach a statistically significant difference (P>0.05 vs vehicle-treated tMCAO group, n=6). The blood flow in the contralateral hemisphere remains unchanged during and after the tMCAO (P>0.05, vs before tMCAO, n=6).

Effects of Xyl-B treatment on brain hemodynamics. Representative images of the hemodynamics are shown (n=6 mice/group).
Xyl-B improved the neurological outcome
We assessed the effects of the Xyl-B treatment on the ischemia-induced neurological deficits using a 4-tiered scoring system19. Although this neurological deficiency assessment is variable, it can indicate the successful establishment of the MCAO model18. As shown in Table 4, the Xyl-B treatment significantly improved the neurological outcome of the tMCAO mice compared with that of the vehicle group (P<0.05 vs Vehicle controls, n=7 mice/group). Consistently, the tMCAO mice that were treated with edaravone also displayed significant neuroprotective effects compared with the vehicle control group (P<0.05 vs Vehicle controls, n=7 mice/group). There was no significant difference in the neurological outcome between the Xyl-B and edaravone groups (P>0.05).
Xyl-B decreased the ROS level and increased the SOD2 level
Reactive oxygen species (ROS) and reactive nitrogen species (RNS), such as nitric oxide (NO), play a key role in the pathophysiological response of the brain after an ischemia injury20. Here, we detected the total ROS levels by DHE staining. As shown in Figure 3A, the total ROS level in the brain tissues from the Xyl-B-treated tMCAO mice decreased compared with that in brain tissues from vehicle-treated tMCAO mice.

Xyl-B treatment reduced ROS generation and iNOS protein expression and increased MnSOD protein expression. (A) Representative images of brain tissues stained with DHE dye (×40), a ROS indicator. Xyl-B treatment refers to a single dose of 50 mg/kg Xyl-B given immediately after acute ischemia. The fluorescence intensity of DHE in the ischemic brain with Xyl-B (50 mg/kg) treatment immediately after tMCAO decreased compared with that in the vehicle group (n=6 mice /group). Western blot analyses of MnSOD (B) and iNOS (C) were performed in the brain tissues from the sham-operated mice, vehicle-treated mice and Xyl-B-treated tMCAO mice. **P<0.01 vs sham-operated controls. ##P<0.01 vs vehicle controls, n=6 mice/group.
To investigate whether Xyl-B affects endogenous antioxidant enzymes, we examined the expression of manganese superoxide dismutase (MnSOD or SOD2). As shown in Figure 3B, the Xyl-B treatment markedly enhanced the protein levels of MnSOD in the brain tissues that were reduced by the tMCAO.
Excessive NO in a brain with an ischemia injury is mainly produced by inducible NO synthase (iNOS)20. The western blotting results showed that the Xyl-B treatment significantly reduced the expression of the iNOS protein in the ischemic brains compared with that in the brains of the vehicle-treated tMCAO group (Figure 3C).
Xyl-B reduced brain inflammatory cytokine mRNA levels
The inflammatory response plays a key role in the pathogenesis of ischemic stroke21. Inflammatory cytokines, including IL-1β, TNF-α, IL-6 and IFN-γ, were detected by quantitative real-time RT-PCR. As shown in Figure 4, these proinflammatory cytokines increased significantly in the vehicle-treated tMCAO mice compared with those in the sham-operated mice. By contrast, the Xyl-B treatment significantly reduced the mRNA expression of IL-1β, TNF-α, IL-6 and IFN-γ.

Inflammatory cytokine mRNA levels in the brain were reduced in the Xyl-B-treated mice. Quantitative real-time RT-PCR was performed to analyze the cytokine mRNA levels, including IL-1β (A), IL-6 (B), TNF-α (C), and IFN-γ (D), in the brain tissues from the sham-operated mice, vehicle-treated mice and Xyl-B-treated tMCAO mice. **P<0.01 vs Sham-operated controls. ##P<0.01 vs Vehicle controls. n=6 mice/group.
Xyl-B protected the blood-brain barrier (BBB)
To evaluate the effects of Xyl-B on BBB permeability after an ischemic injury, BBB leakage was measured by Evans blue extravasation. As shown in Figure 5, the Evans blue extravasation index in the ipsilateral/contralateral hemisphere was significantly increased in the tMCAO group compared with that in the sham group, indicating a BBB disruption, while it was significantly decreased by the Xyl-B and edaravone treatments, indicating that Xyl-B protected BBB integrity after an ischemic injury.

Evans blue extravasation was attenuated in the Xyl-B-treated tMCAO mice. Evans blue extravasation was used to determine BBB disruption in the brains of the sham-operated mice, vehicle-treated mice and Xyl-B-treated tMCAO mice. Evans blue extravasation index of the ipsilateral/contralateral hemisphere is shown in the bar graph. **P<0.01 vs sham-operated controls. ##P<0.01 vs vehicle controls. n=6 mice/group.
Xyl-B suppressed the expression of TLR4 and the activation of NF-κB
The TLR4-NF-κB signaling pathway is enhanced in the brain after the tMCAO and, consequently, leads to inflammatory responses. As shown in Figure 6, the western blotting data confirmed that the protein expression of TLR4 and NF-κB significantly increased after ischemia. The Xyl-B treatment significantly decreased the ischemia-induced elevation in the expression of TLR4 and NF-κB in the ipsilateral hemisphere of the tMCAO mice compared with that in the corresponding vehicle group (Figure 6A & B). Consistent with the Western blot data, the immunohistochemistry staining of the mouse brain preparations showed that Xyl-B greatly attenuated the elevation of NF-κB protein expression that was induced by tMCAO (Figure 6C). In the basal state, NF-κB is stabilized in the cytoplasm, and the distribution of NF-κB from the cytoplasm to the nucleus indicates the activation of NF-κB upon stimulation. The present data demonstrated that the nuclear translocation of NF-κB p65 was enhanced after ischemia, which was significantly reduced by the Xyl-B treatment (Figure 6D).

Xyl-B treatment reduced the protein expression levels of TLR4 and NF-κB and repressed the activation of NF-κB. Western blot analysis of TLR4 shows that the protein expression levels of TLR4 (A) and NF-κB (B) were reduced in the Xyl-B-treated mice vs those in the vehicle controls. (C) Representative immunohistochemistry staining images (x40) demonstrate that the expression of NF-κB is reduced in the Xyl-B-treated mice vs that in the vehicle controls. “C” and “I” represent the contralateral hemisphere and ipsilateral hemisphere, respectively. (D) Western blot analysis was used to determine the expression of NF-κB p65 in the cytoplasmic and nuclear fractions isolated from the ipsilateral hemisphere of the sham-operated mice, vehicle-treated mice and Xyl-B-treated tMCAO mice. *P<0.01 vs sham-operated controls. ##P<0.01 vs vehicle controls. n=6 mice/group.
Discussion
The main findings of this study are as follows: (1) Multiple administrations of Xyl-B at 12.5, 25 or 50 mg/kg before ischemia significantly reduces brain infarct volume in a dose-dependent manner; (2) a single dose of Xyl-B at 50 mg/kg given after the onset of ischemia significantly reduces brain infarct volume, and the benefit is still significant at 2 h of ischemia, and the maximum benefit is reached immediately after ischemia; (3) a single dose of Xyl-B given immediately after ischemia (Xyl-B treatment) markedly improves neurological outcomes and protects the integrity of the BBB but does not cause a significant hemodynamic change after ischemia; (4) Xyl-B treatment dramatically reduced ROS overproduction and increased the reduction in MnSOD induced by acute ischemia; (5) the Xyl-B treatment significantly decreased the mRNA levels of proinflammatory cytokines, such as IL-1β, TNF-α, IL-6 and IFN-γ, after ischemic brain injury; and (6) Xyl-B treatment attenuates inflammatory protein expression levels, including TLR4, NF-κB and iNOS, after ischemia.
Pharmacological therapies for acute ischemic strokes require further development. Tissue plasminogen activator (tPA) is the only approved drug for the treatment of acute ischemic strokes, but it has the limitation of a short therapeutic window and some adverse side effects22. The marine compound Xyl-B has a unique chemical structure and was isolated from mangrove fungus Xylaria sp8. It was found that Xyl-B protected PC-12 cells against oxygen glucose deprivation (OGD) and 1-methyl-4-phenylpyridinium (MPP+)-induced damage10,11. Recently, we found that pretreatment with Xyl-B reduced brain damage in neonatal mouse hypoxic-ischemic injury17. However, the effects of Xyl-B on adult ischemic brain injuries had not been previously investigated. In this study, we found that both pretreatment and post-treatment with Xyl-B after the onset of ischemia exerted significant protective and therapeutic effects on tMCAO-induced stroke mice. Our findings provide important evidence regarding the potential application of Xyl-B in stroke therapy with a therapeutic window of up to 2 h after the onset of ischemia with single-dose administration. Our previous study demonstrated that Xyl-B increased endothelial NO generation in human umbilical vein endothelial cells9, indicating that Xyl-B may influence hemodynamics via vasodilation. In this study, Xyl-B did not significantly change brain hemodynamics in the tMCAO mice, suggesting that Xyl-B may provide protective effects through other mechanisms rather than through improving the blood supply in the ischemic brain.
Excessive ROS generation and the increased expression or activity of iNOS under ischemic conditions in stroke may result in an increase in NO production and, subsequently, an increase in peroxynitrite formation23. Our previous studies suggested that Xyl-B inhibited ROS or RNS generation through multiple approaches, such as the suppression of NADPH oxidase and the activation of endogenous antioxidants9,10,13. Thus, the protective effects of Xyl-B against a stroke might be through inhibiting ischemia-induced oxidative stress. In this study, we found that the Xyl-B treatment attenuated ROS generation in the ischemic brains of the tMCAO mice and reduced the iNOS protein expression, suggesting that it would decrease peroxynitrite formation. Edaravone, a novel free-radical scavenger, was used to treat the tMCAO mice in parallel with Xyl-B, and it reduced brain infarction in tMCAO mice. Thus, our results suggested that treatment with Xyl-B can suppress excessive ROS generation after cerebral ischemia. Our previous studies have demonstrated that Xyl-B can increase some endogenous antioxidant enzymes and proteins, such as GSH, HO-1 and Bcl-29,13. Here, we found that Xyl-B treatment significantly enhanced the reduction of MnSOD after ischemia. MnSOD is one of the major endogenous antioxidant enzymes and is considered an effective therapeutic target for the treatment of cerebral ischemia24. MnSOD mimics have been shown to be neuroprotective after acute ischemia25. Altogether, the findings of this study suggest that the suppression of excessive ROS generation and the elevation of endogenous antioxidants by Xyl-B was involved in its therapeutic effects against acute cerebral ischemia.
Multiple lines of evidence, including experimental and clinical studies, indicate that an acute and prolonged inflammatory response plays a key role in ischemic brain injury26,27,28. Cytokines, such as IL-1β, TNF-α, IL-6 and IFN-γ, have been demonstrated to increase 24 h after ischemia in a tMCAO model, and the reduction in these cytokines was beneficial for the therapy of ischemic strokes18. Consistently, this study also found that IL-1β, TNF-α, IL-6 and IFN-γ mRNA expression increased after ischemia in tMCAO stroke mice, while expression was reduced by Xyl-B treatment, indicating that Xyl-B attenuated inflammatory responses both during and after acute ischemic brain injury. However, ischemia and ischemia-associated inflammation lead to higher BBB permeability and disruption29. The inhibition of cytokines, such as TNF-α, preserved BBB integrity after ischemia insult30. Xyl-B treatment significantly reduced ischemia-induced BBB disruption as indicated by the Evans blue extravasation assay, suggesting that Xyl-B may maintain BBB integrity by reducing the generation of inflammatory cytokines.
It has been revealed that the TLR4-NF-κB signaling pathway is critical for activating secondary inflammation in strokes5,6,31. TLR4 can signal through adaptor proteins, such as myeloid differentiation primary-response protein 88 (MyD88), to activate NF-κB, in turn resulting in the elevation of inflammatory cytokines31. Animals deficient in TLR4 show significantly reduced infarct sizes in experimental stroke models32. Consistently, we confirmed that ischemia induced TLR4 expression and the nuclear translocation of NF-κB, whereas Xyl-B markedly attenuated the activation of TLR4 and NF-κB signaling after ischemia. Similarly, Xyl-B also significantly reduced the downstream genes of NF-κB, TNF-α and IL-1β after ischemic injury. Our present findings are the first to demonstrate that Xyl-B attenuates the activation of the TLR4-NF-κB signaling pathway and inflammatory responses under diseased conditions.
The activation of NF-κB and the excessive generation of inflammatory cytokines is also closely related to increased iNOS expression33, while the inhibition of iNOS expression could reduce ischemic brain injury34. Xyl-B treatment can decrease the expression of iNOS and inflammatory cytokines; thus, the suppression of iNOS-mediated inflammatory responses may be involved in the therapeutic benefit of Xyl-B. Lipopolysaccharide (LPS) is a stimulator of the TLR4-NF-κB inflammatory signaling pathway35, and it was found that TRPM7 is involved in LPS-induced ROS generation in endothelial cells and increased inflammation in aldosterone-infused mice36. Because Xyl-B also blocks TRPM7 currents14, it is very possible that Xyl-B may attenuate ischemia-induced inflammatory responses by inhibiting TRPM7. However, future studies are needed to investigate these potential mechanisms.
In summary, a single injection of 50 mg/kg of the marine compound Xyl-B within 2 h after tMCAO significantly reduces the infarct volume without affecting cerebral blood flow. This study also demonstrated that Xyl-B offers protection against focal ischemic strokes by reducing excessive ROS production, increasing the antioxidant MnSOD levels and inhibiting TLR4-NF-κB–mediated inflammatory reactions. The findings of this study indicate that Xyl-B is a potential new drug candidate for the treatment of strokes.
Author contribution
Guan-lei WANG and Wen-liang CHEN designed the research study; Ni PAN, Liu-yi LU, Jian-wen CHEN and Li-yan ZHAO performed the research study; Mei LI and Guo-hao WANG analyzed the data; Fang-yun SUN, Hong-shuo SUN, Ji-yan PANG, Jian-ding CHENG, Jie LIU, and Yong-yuan GUAN revised the manuscript. Wen-liang CHEN and Guan-lei WANG wrote the paper. Hong-shuo SUN and Xue-jun WEN helped with polishing the language of the manuscript.
References
Davis SM, Donnan GA, Parsons MW, Levi C, Butcher KS, Peeters A, et al. Effects of alteplase beyond 3h after stroke in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET):a placebo-controlled randomised trial. Lancet Neurol 2008; 7: 299–309.
Simats A, Garcia-Berrocoso T, Montaner J . Neuroinflammatory biomarkers: from stroke diagnosis and prognosis to therapy. Biochim Biophys Acta 2016; 1862: 411–24.
Tobin MK, Bonds JA, Minshall RD, Pelligrino DA, Testai FD, Lazarov O . Neurogenesis and inflammation after ischemic stroke: what is known and where we go from here. J Cereb Blood Flow Metab 2014; 34: 1573–84.
Jin R, Yang G, Li G . Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukocyte Biol 2010; 87: 779–89.
Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010; 171: 258–67.
Belinga VF, Wu GJ, Yan FL, Limbenga EA . Splenectomy following MCAO inhibits the TLR4-NF-kappaB signaling pathway and protects the brain from neurodegeneration in rats. J Neuroimmunol 2016; 293: 105–13.
Shih RH, Wang CY, Yang CM . NF-kappaB signaling pathways in neurological inflammation: a mini review. Frontiers Mol Neurosci 2015; 8: 77.
Lin Y, Wu X, Feng S, Jiang G, Luo J, Zhou S, et al. Five unique compounds: xyloketals from mangrove fungus Xylaria sp from the South China Sea coast. J Org Chem 2001; 66: 6252–6.
Chen WL, Qian Y, Meng WF, Pang JY, Lin YC, Guan YY, et al. A novel marine compound xyloketal B protects against oxidized LDL-induced cell injury in vitro. Biochem Pharmacol 2009; 78: 941–50.
Zhao J, Li L, Ling C, Li J, Pang JY, Lin YC, et al. Marine compound Xyloketal B protects PC12 cells against OGD-induced cell damage. Brain Res 2009; 1302: 240–7.
Lu XL, Yao XL, Liu Z, Zhang H, Li W, Li Z, et al. Protective effects of xyloketal B against MPP+-induced neurotoxicity in Caenorhabditis elegans and PC12 cells. Brain Res 2010; 1332: 110–9.
Li S, Shen C, Guo W, Zhang X, Liu S, Liang F, et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson's disease models. Marine Drugs 2013; 11: 5159–89.
Li ZX, Chen JW, Yuan F, Huang YY, Zhao LY, Li J, et al. Xyloketal B exhibits its antioxidant activity through induction of HO-1 in vascular endothelial cells and zebrafish. Marine Drugs 2013; 11: 504–22.
Chen WL, Turlova E, Sun CL, Kim JS, Huang S, Zhong X, et al. Xyloketal B suppresses glioblastoma cell proliferation and migration in vitro through inhibiting TRPM7-regulated PI3K/Akt and MEK/ERK signaling pathways. Marine Drugs 2015; 13: 2505–25.
Zhao LY, Li J, Yuan F, Li M, Zhang Q, Huang YY, et al. Xyloketal B attenuates atherosclerotic plaque formation and endothelial dysfunction in apolipoprotein e deficient mice. Marine Drugs 2015; 13: 2306–26.
Zeng Y, Guo W, Xu G, Wang Q, Feng L, Long S, et al. Xyloketal-derived small molecules show protective effect by decreasing mutant Huntingtin protein aggregates in Caenorhabditis elegans model of Huntington's disease. Drug Design Development Ther 2016; 10: 1443–51.
Xiao AJ, Chen W, Xu B, Liu R, Turlova E, Barszczyk A, et al. Marine compound xyloketal B reduces neonatal hypoxic-ischemic brain injury. Marine Drugs 2015; 13: 29–47
Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y . CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 2013; 44: 190–7.
Bederson JB, Pitts LH, Miles T, Nishimura MC, David RL, Henry B . Rat middle cerebral artery occlusion: evaluation of the model and development of a neuroligic examination. Stroke 1986; 17: 472–6.
Huang YY, Huang XQ, Zhao LY, Sun FY, Chen WL, Du JY, et al. ClC-3 deficiency protects preadipocytes against apoptosis induced by palmitate in vitro and in type 2 diabetes mice. Apoptosis 2014; 19: 1559–70.
Sun YY, Li Y, Wali B, Lee J, Heinmiller A, Abe K, et al. Prophylactic edaravone prevents transient hypoxic-ischemic brain injury: implications for perioperative neuroprotection. Stroke 2015; 46: 1947–55.
Yu S, Liu X, Shen Y, Xu H, Yang Y, Ding F . Therapeutic benefits of combined treatment with tissue plasminogen activator and 2-(4-methoxyphenyl)ethyl-2-acetamido-2-deoxy-beta-d-pyranoside in an animal model of ischemic stroke. Neuroscience 2016; 327: 44–52.
Wang T, Gu J, Wu PF, Wang F, Xiong Z, Yang YJ, et al. Protection by tetrahydroxystilbene glucoside against cerebral ischemia: involvement of JNK, SIRT1, and NF-kappaB pathways and inhibition of intracellular ROS/RNS generation. Free Radical Biol Med 2009; 47: 229–40.
Kim GW, Kondo T, Noshita N, Chan PH . Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice. Implications for the production and role of superoxide radicals. Stroke 2002; 33: 809–15.
Huang HF, Guo F, Cao YZ, Shi W, Xia Q . Neuroprotection by manganese superoxide dismutase (MnSOD) mimics: antioxidant effect and oxidative stress regulation in acute experimental stroke. CNS Neurosci Ther 2012; 18: 811–8.
Wang Q, Tang XN, Yenari MA . The inflammatory response in stroke. J Neuroimmunol 2007; 184: 53–68.
Emsley HC, Hopkins SJ . Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol 2008; 7: 341–53.
McColl BW, Allan SM, Rothwell NJ . Systemic infection, inflammation and acute ischemic stroke. Neuroscience 2009; 158: 1049–61.
Chelluboina B, Klopfenstein JD, Pinson DM, Wang DZ, Vemuganti R, Veeravalli KK . Matrix metalloproteinase-12 induces blood-brain barrier damage after focal cerebral ischemia. Stroke 2015; 46: 3523–31.
Abdullah Z, Rakkar K, Bath PM, Bayraktutan U . Inhibition of TNF-alpha protects in vitro brain barrier from ischaemic damage. Mol Cell Neurosci 2015; 69: 65–79.
Zhang J, Fu B, Zhang X, Chen L, Zhang L, Zhao X, et al. Neuroprotective effect of bicyclol in rat ischemic stroke: down-regulates TLR4, TLR9, TRAF6, NF-kappaB, MMP-9 and up-regulates claudin-5 expression. Brain Res 2013; 1528: 80–8.
Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I . Toll-Like receptor 4 is involved in subacute stress–induced neuroinflammation and in the worsening of experimental stroke. Stroke 2008; 39: 1314–20.
Ahn KS, Aggarwal BB . Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann New York Acad Sci 2005; 1056: 218–33.
Anthony Jalin AM, Lee JC, Cho GS, Kim C, Ju C, Pahk K, et al. Simvastatin reduces lipopolysaccharides-accelerated cerebral ischemic injury via inhibition of nuclear factor-kappa B activity. Biomol Ther 2015; 23: 531–8.
Sarmiento D, Montorfano I, Caceres M, Echeverria C, Fernandez R, Cabello-Verrugio C, et al. Endotoxin-induced vascular endothelial cell migration is dependent on TLR4/NF-kappaB pathway, NAD(P)H oxidase activation, and transient receptor potential melastatin 7 calcium channel activity. Int J Biochem Cell Biol 2014; 55: 11–23.
Sontia B, Montezano AC, Paravicini T, Tabet F, Touyz RM . Downregulation of renal TRPM7 and increased inflammation and fibrosis in aldosterone-infused mice: effects of magnesium. Hypertension 2008; 51: 915–21.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No 81370897, 81441115, 21172271, 81402926, 81430046), the NSFC-CIHR Joint Health Research Initiative Proposal (No 81361128011), the Canadian Institutes of Health Research CIHR-NSFC China-Canada Joint Health Research Initiative (CIHR, FRN #132571), the Guangdong Provincial Department of Science and Technology (No 2016A050502023), the National Key New Drug Creation Program (No 2009ZX09103-039), the Research Funds for Provincial Key Laboratory from the Department of Education of Guangdong Province (No 50000-3211105), and the Youth Program from Guangzhou City Bureau of Education (No 2012C204).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Pan, N., Lu, Ly., Li, M. et al. Xyloketal B alleviates cerebral infarction and neurologic deficits in a mouse stroke model by suppressing the ROS/TLR4/NF-κB inflammatory signaling pathway. Acta Pharmacol Sin 38, 1236–1247 (2017). https://doi.org/10.1038/aps.2017.22
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.22
Keywords
This article is cited by
-
Selenium Deficiency Causes Iron Death and Inflammatory Injury Through Oxidative Stress in the Mice Gastric Mucosa
Biological Trace Element Research (2024)
-
Waixenicin A, a marine-derived TRPM7 inhibitor: a promising CNS drug lead
Acta Pharmacologica Sinica (2020)
-
Effects of nicorandil on neurobehavioral function, BBB integrity, edema and stereological parameters of the brain in the sub-acute phase of stroke in a rat model
Journal of Biosciences (2020)
-
The antidepressant effects of asperosaponin VI are mediated by the suppression of microglial activation and reduction of TLR4/NF-κB-induced IDO expression
Psychopharmacology (2020)
-
Costunolide alleviates HKSA-induced acute lung injury via inhibition of macrophage activation
Acta Pharmacologica Sinica (2019)
