
Abstract
Stroke is one of the major causes of mortality and morbidity worldwide, yet novel therapeutic treatments for this condition are lacking. This review focuses on the roles of the transient receptor potential melastatin 2 (TRPM2) ion channels in cellular damage following hypoxia-ischemia and their potential as a future therapeutic target for stroke. Here, we highlight the complex molecular signaling that takes place in neurons, glial cells and the blood-brain barrier following ischemic insult. We also describe the evidence of TRPM2 involvement in these processes, as shown from numerous in vitro and in vivo studies that utilize genetic and pharmacological approaches. This evidence implicates TRPM2 in a broad range of pathways that take place every stage of cerebral ischemic injury, thus making TRPM2 a promising target for drug development for stroke and other neurodegenerative conditions of the central nervous system.
Similar content being viewed by others


Introduction
Stroke is a complex neurological condition that causes irreparable destruction of brain tissue. In 2013, stroke was the 5th leading cause of death in the United States, where it accounted for 1 out of every 20 deaths and killing as many as 130 000 people each year1. While recent advances in research have identified several potential mechanisms underlying neuronal death following stroke, the treatments for this condition remain limited. It has been estimated that by 2050, the incidence of stroke will more than double, and by 2030, total stroke-related direct medical costs will rise from $71.6 billion to $184.1 billion1. Therefore, there is an urgent need for novel therapeutic opportunities beyond the current stroke treatments.
The current understanding of stroke pathology revolves around the biochemical cascade that begins with ischemia and lasts long after blood flow is restored2,3. Energy failure and ATP depletion due to low oxygen levels lead to a massive release of glutamate into the synaptic cleft and to excessive Ca2+ influx through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-Methyl-D-aspartic acid (NMDA) receptors. As a result, pro-apoptotic signaling and reactive oxygen species production are activated2,3. The severity of the ischemic insult dictates the extent of further neuronal cell death, activation of inflammatory cascades and prolonged apoptosis3. A previous and popular strategy was pharmacological targeting of the activation of NMDA receptors after the excessive glutamate release following ischemia. Several compounds, including dizocilpine maleate (MK-801)4,5, aptiganel hydrochloride (Cerestat)6, dextromethorphan (DMX)7 and CGS 19755 (Selfotel)8,9 showed promising results in rodent models. However, by 2001, all clinical trials using NMDA receptor antagonists as treatment for stroke were deemed unsuccessful10,11,12,13,14,15. Therefore, effort was re-focused on finding novel, non-glutamate therapeutic targets for hypoxic-ischemic cell death.
Non-glutamate targets for stroke therapy
Several non-glutamate ion channels have been identified as potential therapeutic targets for cerebral ischemia in rodent models, including the following examples.
1) Acid-sensing ion channels. The ASIC1a channel is widely expressed in the brain, and its activation has been shown to contribute to neuronal cell death in both in vitro and in vivo models16,17.
2) Volume-regulated anion channels. Cerebral edema and swelling are some of the factors contributing to delayed neuronal death following cerebral ischemia. Volume-regulated anion channels (VRACs) regulate cell volume via efflux of ions that are followed by water18. In astrocytes, VRACs have been shown to conduct excitatory amino acids (EAAs), aspartate and glutamate, after ischemia, thus contributing to excitotoxicity19,20,21.
3) Hemichannels. Hemichannels, proteins involved in the formation of gap junctions, have also been proposed as non-glutamate contributors of neuronal death in ischemia22. They have been implicated in contributing to anoxic depolarization, leading to cell death in the penumbra region, as well as efflux of vital nutrients from neurons, thus worsening the effects of energy failure caused by low-oxygen conditions23,24,25.
4) Transient Receptor Potential Melastatin (TRPM) subfamily. Transient Receptor Potential melastatin (TRPM) channels are calcium-permeable, ubiquitously expressed cation channels26,27. TRPM7 has recently emerged as one of the major contributors to non-glutamate-induced cell death following ischemia28,29,30. In vitro pharmacological inhibition31 and in vivo siRNA suppression of TRPM7 in rodents32 were shown to significantly reduce neuronal death. Moreover, conditions such as low pH and reactive oxygen species were found to enhance TRPM7 activity33,34. The contribution of a closely related TRPM family member, TRPM2, to hypoxic-ischemic brain injury has also been investigated.
In this review, we focus on the role of TRPM2 in neuronal and non-neuronal mechanisms that contribute to the devastating effects of cerebral ischemia.
TRPM2: structure and biophysical properties
TRPM2, the second member of the melastatin subfamily of the transient receptor potential (TRP) channel superfamily, is a calcium-permeable, non-selective cation channel35. It is broadly expressed within the CNS, heart, lung, liver and pancreas36. At the cellular level, TRPM2 has been identified in multiple cell types, including neurons37,38,39,40,41,42, microglia43,44,45,46,47,48,49, astrocytes50, macrophages51,52, neutrophils53,54,55, dendritic cells56, megakaryocytes57, endothelial vascular cells58,59,60,61,62, cardiomyocytes63 and pancreatic β-cells64,65. The ubiquitous distribution of TRPM2 indicates that it may play roles in a wide range of physiological processes. In addition to its role as a plasma membrane channel, TRPM2 has been shown to also be localized to the lysosomal compartment, where it regulates calcium mobilization from intracellular compartments and contributes to H2O2-induced apoptosis of β cells66.
The human TRPM2 gene is located on chromosome 21q22.3, spanning approximately 90 kb and encoding 1503 amino acid residues67. TRPM2 has also been cloned from mouse and rat tissues, encoding 1507 amino acid residues, with a predicted molecular weight of 172 kDa and 83%–85% similarity to human TRPM2 at the nucleotide and protein levels, repectively68,69. Molecularly, the TRPM2 channel is composed of four identical subunits, each consisting of a 730-amino acid N terminus with an IQ-like calmodulin-binding motif (amino acids 406-416)70, 6 transmembrane domains (S1-S6) with a pore-forming loop located between S5 and S6, and a C terminus containing a highly conserved TRP box, a coiled-coil domain and a unique adenosine diphosphate ribose (ADPR) pyrophosphatase NUDT9-H domain (amino acids 1197-1503) (Figure 1)71. The NUDT9-H domain contains an 11-residue ADPR binding pocket72; TRPM2 has been shown to be gated by free ADPR71. A site-directed mutagenesis study identified that hydrogen bonding of Arg1433 and Tyr1349 is necessary for TRPM2 activation by ADPR73. The enzymatic activity of NUDT9-H is not required for channel gating74, however it plays a role in TRPM2 surface expression75. When expressed on its own, the NUDT9-H domain also has measurable enzymatic activity, thus making TRPM2 a “chanzyme”. However, the specific role of that activity remains to be defined71. In addition to the NUDT9-H domain, the C-terminus contains a coiled-coil domain that was shown to be critical in mediating the tetrameric assembly of the channel76. The N-terminal IQ-like motif is important in activating TRPM2 current by intracellular Ca2+ in an ADPR-free manner70,77,78,79. In addition to full-length TRPM2 (TRPM2-L), several splice isoforms with varying degrees of activity have been identified: TRPM2-ΔN80, TRPM2-ΔC80, TRPM2-S59, TRPM2-SSF81 and TRPM2-TE82.

TRPM2 protein structure and modulators of TRPM2 activity. Extracellular agents such as H2O2, ROS, TNFα, Aβ and concanavalin A enhanced TRPM2 activity via production of intracellular ADP that gates TRPM2 via binding to the NUDT9-H domain. AMP and 8-Br-cADPR reduce TRPM2 activity through interaction with the NUDT9-H domain. Protons and divalent heavy metal cations reduce TRPM2 activity through interaction with the pore region. Ca2+ gates TRPM2 via CaM interaction with N-terminal IQ-like motif. Structurally unrelated compounds, such as FFA, clotrimazole, 2-APB, ACA, scalaradial and AG490, 555 and 556, are also able to inhibit TRPM2, although their mechanisms of action remain to be elucidated.
Upon activation, TRPM2 displays a linear current-voltage (I-V) relationship, with a single-channel conductance range of 52-60 pS at negative potentials and ∼72 pS at positive potentials69,71,79,83,84. TRPM2 is permeable to Na+, K+, Ca2+ and Mg2+. The relative permeabilities of TRPM2 to these ions are reported as PK/PNa – 1.1, PCa/PNa – 0.9 and PMg/PNa – 0.585. Several key residues identified within the pore-forming region dictate channel function as well as conductance of certain ions. The two conserved cysteine residues, C996 and C1008, were shown to be critical for TRPM2 gating by ADPR and voltage86. The point mutation P1018L87, present in Guamanian amyotrophic lateral sclerosis and parkinsonism-dementia patients, produces a fast-desensitizing channel. The permeability of TRPM2 to Ca2+ and Mg2+ was shown to be controlled by four residues within the pore-forming loop: E960, Q981, D987 and E102285.
Several extracellular stimuli, including reactive oxygen species88, H2O280, amyloid β-peptide89, concanavalin A90, tumor necrosis factor-α91 and zinc ions45, have been shown to induce TRPM2 activation via metabolic production of intracellular ADPR. While ADPR is considered to be the most potent TRPM2 activator (EC50 of 10–80 μmol/L)71,92, there has been much controversy around other proposed TRPM2 activators. Several studies suggested that other nucleotides, such as 2′-O-acetyl-ADPr (OAADPr), cyclic ADP (cAPDr), nicotinamide-adenine dinucleotide (NAD), nicotinic acid-adenine dinucleotide (NAAD), and NAAD-phosphate (NAADP), can also enhance TRPM2 activity93,94. However, a recent study showed, using affinity-purified-specific ADPR hydrolase to purify commercially available pyridine dinucleotides, that NAD, NAAD and NAADP were incapable of stimulating TRPM2 activity, even at concentrations substantially higher than cytosolic. Instead, they identified ADPR-2′-phosphate (ADPRP) as a direct TRPM2 agonist95. Full activation of TRPM2 is highly dependent on the presence of intracellular and/or extracellular Ca2+; ADPR-induced TRPM2 current was shown to be significantly reduced in the absence of Ca2+. It has been proposed that intracellular calcium sensitizes TRPM2 to ADPR via calcium-dependent interaction of calmodulin (CaM) with the N-terminal IQ-like motif. Calcium can also gate the channel in the absence of ADPR, with an EC50 of 17 μmol/L70,77,78,79.
In addition to agonists, several non-specific inhibitors of TRPM2 have been described. Adenosine monophosphate (AMP) inhibits the channel activity, potentially via binding to the NUDT9-H domain with IC50 values of 10 μmol/L and 70 μmol/L for endogenous and recombinant channels, respectively53,92,96. 8-Bromo-cyclic inosine diphosphoribose (8-Br-cADPR, IC50 100 μmol/L) was shown to inhibit TRPM2 gating by cADPR and H2O292. Protons97,98,99 and divalent heavy metal cations100,101,102 also caused TRPM2 inhibition by targeting the extracellular pore region. Several structurally unrelated pharmacological agents have been identified as TRPM2 inhibitors. Those include flufenamic acid (FFA, IC50 50–1000 μmol/L)103, the anti-fungal agents clotrimazole and econazole (IC50 3–30 μmol/L)104, 2-aminoethoxydiphenyl borate (2-APB, IC50 1.2 μmol/L)105, N-(p-amylcinnamoyl) anthranilic acid (ACA, IC50 1.7 μmol/L)106, tyrphostin AG-related compounds (AG490, AG555 and AG556)107,108 and the marine-derived compounds scalaradial and 12-deacetylscalaradial (IC50 210 nmol/L)109. It is important to note that these compounds affect a wide variety of ion channels and proteins, and none of them are selective for TRPM2. Therefore, efforts should be made to develop TRPM2-specific inhibitors in order to further elucidate the physiological functions of this channel.
In addition to nucleotides and pharmacological agents, TRPM2 activity was also shown to be modulated via interactions with other proteins. As mentioned above, CaM-TRPM2 interaction at the N-terminal IQ-like motif facilitates TRPM2 activation by Ca2+. Mutation of the IQ-like motif or expression of a CaM mutant that is unable to bind Ca2+ significantly inhibits the rate of development of H2O2-induced TRPM2 Ca2+ conductance70,79. The non-receptor protein tyrosine phosphatase PTPL1 directly interacts with TRPM2 and reduces TRPM2 phosphorylation, Ca2+ influx and cell death induced by H2O2 and TNF-α in the human monocytic cell line U937110.
Neuronal TRPM2 in cerebral ischemia
Neuronal cell death is the hallmark of ischemic insult resulting in life-long, debilitating and irreversible consequences for survivors. TRPM2 is broadly expressed in neurons. Primary rat cortical cultures exposed to H2O2 undergo rapid apoptotic cell death; treating these neurons with TRPM2 siRNA significantly inhibits H2O2-induced intracellular Ca2+ influx and neuronal cell death41. This indicates that neuron-specific TRPM2 may contribute to the pathology of cerebral ischemia. CA1 hippocampal neurons are highly vulnerable to oxidative stress, and ischemic injury often causes irreparable damage to the hippocampus. CA1 neurons from rat hippocampal slices showed H2O2-induced inward current that was inhibited by the TRPM2 antagonist clotrimazole111. Moreover, activation of TRPM2-like currents in these neurons required concomitant activation and Ca2+ influx via voltage-gated Ca2+ channels and NMDARs, the two events that take place following hypoxia-ischemia111. Another study demonstrated that TRPM2-deficient CA1 pyramidal neurons were resistant to increases in cytosolic Zn2+ concentrations, thus implicating TRPM2 in delayed neuronal cell death post-ischemia112.
The contribution of TRPM2 to ischemic cell death has also been addressed in several animal models of hypoxia-ischemia. Compared to wild-type mice, TRPM2-null mice subjected to transient middle cerebral artery occlusion (tMCAO) exhibited a reduction of approximately 40% in infarct volumes39. However, when TRPM2-null mice were subjected to permanent MCAO (pMCAO), the infarct severity was comparable to that of wild-type mice. This was hypothesized to be due to the lack of reperfusion following the procedure, thus eliminating the production of H2O2, which is a major activator of TRPM2-like currents. Thus, it is possible that TRPM2-null mice are only resistant to ischemia-reperfusion injury due to reduced vulnerability to H2O2, while under pMCAO conditions, the insult becomes so severe that it outweighs the neuroprotective effects of TRPM2 deletion39. Similarly, hippocampal neurons cultured from these mice showed reduced cell death following one hour of oxygen-glucose deprivation (OGD)39. At the molecular level, the study observed the activation of the pro-survival Akt pathway and the inhibition of downstream glycogen synthase kinase 3β (GSK-3β), thus tipping the scale towards cell survival in TRPM2-null mice39.
Genetic deletion of TRPM2 was also shown to be neuroprotective in a developmental model of hypoxic-ischemic brain damage113. Compared to wild-type littermates, TRPM2+/- and TRPM2-/- neonatal mice had reduced brain infarct volumes, improved sensorimotor outcomes, reduced expression of inflammatory markers and reduced loss of brain mass following hypoxia-ischemia113. At the molecular level, TRPM2+/- and TRPM2-/- neonatal mice showed increased pro-survival signaling, suggesting that genetic knockout of TRPM2 exerts its neuroprotective effects via the Akt/GSK-3β pathway113. These results confirmed the findings from the adult mouse model of tMCAO. Another study used a novel inhibitor of GSK-3β, TDZD-8, to show that deactivation of GSK-3β via phosphorylation on Ser9 is neuroprotective in a neonatal mouse model of hypoxia-ischemia. These findings confirmed the involvement of the Akt/GSK-3β signaling pathway in neuronal survival following ischemic insult114.
The role of neuronal TRPM2 in cerebral ischemia was shown to be sexually dimorphic. Pharmacological inhibition of TRPM2 with ACA106, 2-APB105, clotrimazole (CTZ)104, flufenamic acid103, and TRPM2 shRNA treatment significantly reduced cell death following OGD in neurons from male but not female animals115. Additionally, intrastriatal lentiviral infection with TRPM2 shRNA following middle cerebral artery occlusion (MCAO) resulted in markedly reduced striatal infarct volumes in male but not female mice115. Another study has shown that TRPM2 channels in male, but not female, hippocampal neurons were activated during reperfusion following OGD116. Similarly, inhibition of TRPM2 activity with clotrimazole 30 min after transient global cerebral ischemia due to cardiac arrest reduced CA1 hippocampal neuronal death only in male mice117. Pre-treatment of adult and aged male mice with the TRPM2 inhibitor tat-M2NX resulted in reduced infarct volumes, while no effect was observed in female mice118. These sex differences have been postulated to be due male-specific androgen signaling, and to preferentially enhanced activity of the enzyme poly(ADP-ribose) polymerase-1 (PARP-1) in the male brain following ischemia119. In female mice, androgens were not sufficient to produce TRPM2 activation120. Collectively, these studies indicate that TRPM2 is expressed in neurons, becomes activated under ischemic brain conditions and contributes to cell death in a sexually dimorphic manner.
Non-neuronal TRPM2 in cerebral ischemia
Pathological post-ischemic changes also require the involvement of non-neuronal cells, such as microglia, astrocytes and other immune cells. Microglia, the macrophages of the central nervous system, were previously implicated in pathology following hypoxic-ischemic injury due to their role in generating a range of inflammatory mediators, such as ROS, cytokines, free radicals, glutamate, proteases, nitric oxide (NO) and H2O2121. Lipopolysaccharide (LPS)-activated primary rat microglia had detectable levels of TRPM2 mRNA and exhibited a robust TRPM2-like Ca2+ conductance following the application of H2O247. Similarly, activation of microglia has also been detected following tMCAO injury. In tMCAO rodent model, cortical mRNA levels of TRPM2 increased in a time-dependent manner, peaking at 7 d post-injury, suggesting a contribution to brain damage following ischemia43. Patch-clamp experiments in human C13 microglia and primary rat microglia in a model of H2O2-induced oxidative stress revealed an upregulation of a TRPM2-like conductance, which was reversibly blocked by flufenamic acid43. A recent study using bone marrow chimeric mice demonstrated that Trpm2 deficiency is protective due to TRPM2-mediated regulation of the migratory ability of peripheral immune cells (neutrophils and macrophages) that infiltrate the injury site and exacerbate post-ischemic inflammation48. At the molecular level, microglial activation was shown to be caused by an increase in TRPM2 activity due to generation of ROS and activation of PARP-149. Moreover, this increase in TRPM2 activity was suppressed by inhibition of protein kinase C (PKC) and NADPH oxidase (NOX), as well as proline-rich tyrosine kinase 2β (PYK2) and downstream MEK/ERK signaling49. Therefore, it has been suggested that PKC/NOX-mediated generation of ROS and subsequent activation of PARP-1 lead to activation of microglial TRPM2. Additionally, activation of the PYK2/MEK/ERK pathway downstream of TRPM2 acts as a positive feedback mechanism for further activation of TRPM249. Another study demonstrated that the release of the pro-inflammatory cytokine interleukin-1β from microglia and U937 monocytes occurs due to TRPM2-dependent activation of NLRP3 inflammasomes122,123. These mechanistic findings provide insight into the role of TRPM2 in microglial activation and neuroinflammation.
Astrocytes are another type of glia that undergo molecular and morphological changes in response to CNS insults, such as hypoxia-ischemia124. While the function of activated or reactive astrocytes in stroke remains controversial, it has been demonstrated that reactive astrocytes express the inducible form of nitric oxide synthase (iNOS) following ischemic injury125. This implicates astrocytes in NO production, which contributes to delayed neuronal cell death125. It has also been shown that reactive astrocytes that form a glial scar following brain injury may inhibit the growth of regenerating axons, thus reducing the recovery following injury126. Human astrocytes treated with the TRPM2 inhibitor clotrimazole or transfected with TRPM2 siRNA were reported to show reduced release of inflammatory and neurotoxic factors and downregulated neuroinflammatory signaling, such as the JNK, p38, ERK42/44 and NFĸB pathways, in response to glutathione depletion50. Therefore, it is possible that TRPM2 activity in these cells could contribute at least in part to their deleterious role in brain injury. However, the role of astrocyte-expressed TRPM2 in cerebral ischemic injury is still unclear.
Studies have also linked glial TRPM2 activation, oxidative stress and inflammatory mechanisms to neurodegenerative conditions such as Alzheimer's disease (AD)127. TRPM2 channel activation and subsequent Ca2+ influx due to oxidative stress and depletion of glutathione levels resulted in inflammatory responses in microglia and astrocytes, which may promote and exacerbate neuronal degeneration50. Additionally, it has been shown that in aging cultured hippocampal neurons, TRPM2 currents were enhanced with time, suggesting that TRPM2 may also contribute to neurodegeneration during neuronal senescence40.
Therefore, the current body of literature indicates that non-neuronal TRPM2 may contribute to inflammatory responses in the CNS following ischemic insult and during other neurodegenerative conditions, potentially exacerbating the extent of brain damage.
Role of TRPM2 in the blood-brain barrier in cerebral ischemia
The blood-brain barrier (BBB) is an intricate network of cells that form a functional barrier that separates the CNS from systemic circulation. It is composed of and maintained by a variety of cell types, including pericytes, astrocytes and endothelial cells. Ischemic conditions lead to dysregulation and breakdown of the molecular integrity of the BBB, leading to vasogenic edema and increased permeability to immune cells into the damaged area128. TRP channels have been previously implicated in BBB permeability, and TRPM2 RNA has been detected in primary rat cultures of brain microvessel endothelial cells129. A recent study demonstrated that TRPM2-mediated pericyte autophagy, secondary to stress-induced Y1485 tyrosine nitration of TRPM2, played a critical role in pericyte injury and apoptosis130. Another study confirmed expression of TRPM2 in human pulmonary artery endothelial cell monolayers and demonstrated that H2O2 exposure elicited calcium influx and increased endothelial cell permeability59. This was attenuated by TRPM2 siRNA silencing and overexpression of the isoform TRPM2-S, which interacts with the isoform TRPM2-L and inhibits H2O2-induced calcium influx59. At the molecular level, it has been demonstrated that PARP-1 is strongly activated in endothelial cells, leading to apoptosis. PARP-1 activation has been linked to post-ischemic disruption of BBB, and administration of PARP-1 inhibitors, 3-aminobenzamide and 4-amino-1,8-naphthalamide, in rodents with transient focal ischemia resulted in decreased edema, immune cell infiltration and preservation of endothelial tight junctions131. PARP-1 activation has been previously shown to be required for oxidative stress-induced activation of TRPM2 in DT40 B cells, and PARP-deficient lymphocytes showed no oxidant-induced TRPM2 activation132. A recent study described the role of endothelial cell-expressed TRPM2 in transendothelial migration of polymorphonuclear neutrophils (PMNs)62. It was shown that siRNA-mediated depletion of TRPM2 in endothelial cells led to a reduction in phosphorylated VE-cadherin62, an adhesion molecule that regulates the opening of adherens junctions and facilitates the migration of PMNs across the blood-brain barrier133. Infiltration of PMNs into the ischemic penumbra is one of the hallmarks of post-ischemic inflammation134; therefore, endothelial TRPM2 activation facilitates the secondary brain injury following neutrophil invasion. Moreover, ROS-induced activation of TRPM2 has been implicated in endothelial cell apoptosis61. Application of H2O2 or TNFα has been shown to induce TRPM2-S phosphorylation at Ser39 by PKCα, leading to supra-normal Ca2+ influx, activation of caspase-3 and endothelial cell death61, which can exacerbate the breakdown of endothelial barrier. Therefore, it is possible that TRPM2 channels also contribute to the increased permeability and eventual breakdown of the BBB following ischemia, thus contributing to edema formation, inflammation and cell death, although their role needs to be confirmed by further studies.
Conclusions
While considerable progress has been made in recent years towards elucidating the cellular and molecular pathogenesis of ischemic brain injury, effective and potent treatments for stroke patients are still lacking. There is increasing evidence that TRPM2 regulates a broad range of pathways in neurons, glia and the cells of the BBB, thus contributing to every stage of brain injury development after ischemia (summarized in Figure 2). This evidence makes TRPM2 a promising target for further research and therapeutic development for several reasons. First, broad expression of TRPM2 in CNS and vasculature suggests that TRPM2 inhibition could be more effective at treating ischemic brain injury, compared to conventional therapies. Second, contributions of TRPM2 to different stages of brain injury suggest that therapeutic agents that target TRPM2 activity may have a longer therapeutic window than conventional therapies. Finally, gaining in-depth insight into TRPM2 downstream signaling may lead to development of therapies that specifically target TRPM2 signaling in specific cell types, leading to specialized treatments for different neurodegenerative conditions.

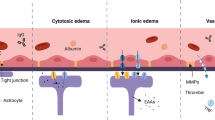
The effects of TRPM2 activation on neurons, glia and the blood-brain barrier under hypoxic/ischemic conditions. TRPM2 activation secondary to oxidative stress and hydrogen peroxide production under ischemic conditions leads to a variety of responses in neurons, glial cells and the cells composing the blood-brain barrier. It has been shown that in neurons, TRPM2 is activated under ischemic conditions and contributes to neuronal cell death, potentially in a sexually dimorphic manner. Current literature indicates that TRPM2 channels may contribute to increased permeability and breakdown of the blood-brain barrier under ischemic conditions. In glial cells, TRPM2 has been shown to mediate the release of neuroinflammatory factors, thus exacerbating the brain damage under ischemic conditions. Together, the evidence indicates that TRPM2 regulates a wide range of pathological events occurring during ischemia, thus making this channel a major target for drug development.
Abbreviations
TRPM2 channel, Transient Receptor Potential Melastatin 2 channel; OGD, oxygen-glucose deprivation; GSK-3β, glycogen synthase kinase 3 beta.
References
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 2016; 133: e38–360.
Iadecola C, Anrather J . The immunology of stroke: from mechanisms to translation. Nat Med 2011; 17: 796–808.
Yenari MA, Han HS . Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci 2012; 13: 267–78.
Buchan AM, Slivka A, Xue D . The effect of the NMDA receptor antagonist MK-801 on cerebral blood flow and infarct volume in experimental focal stroke. Brain Res 1992; 574: 171–7.
Gerriets T, Stolz E, Walberer M, Kaps M, Bachmann G, Fisher M . Neuroprotective effects of MK-801 in different rat stroke models for permanent middle cerebral artery occlusion: adverse effects of hypothalamic damage and strategies for its avoidance. Stroke 2003; 34: 2234–9.
Lees KR . Cerestat and other NMDA antagonists in ischemic stroke. Neurology 1997; 49: S66–9.
van Rijen PC, Verheul HB, van Echteld CJ, Balazs R, Lewis P, Nasim MM, et al. Effects of dextromethorphan on rat brain during ischemia and reperfusion assessed by magnetic resonance spectroscopy. Stroke 1991; 22: 343–50.
Grotta JC, Picone CM, Dedman JR, Rhoades HM, Strong RA, Earls RM, et al. Neuronal protection correlates with prevention of calcium-calmodulin binding in rats. Stroke 1990; 21: III28–III31.
Grotta JC, Picone CM, Ostrow PT, Strong RA, Earls RM, Yao LP, et al. CGS-19755, a competitive NMDA receptor antagonist, reduces calcium-calmodulin binding and improves outcome after global cerebral ischemia. Ann Neurol 1990; 27: 612–9.
Albers GW, Goldstein LB, Hall D, Lesko LM . Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA 2001; 286: 2673–82.
Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF . Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg 1999; 91: 737–43.
Albers GW, Clark WM, Atkinson RP, Madden K, Data JL, Whitehouse MJ . Dose escalation study of the NMDA glycine-site antagonist licostinel in acute ischemic stroke. Stroke 1999; 30: 508–13.
Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, et al. Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke 2000; 31: 347–54.
Davis SM, Albers GW, Diener HC, Lees KR, Norris J . Termination of Acute Stroke Studies Involving Selfotel Treatment. ASSIST Steering Committed. Lancet 1997; 349: 32.
Sacco RL, DeRosa JT, Haley EC Jr, Levin B, Ordronneau P, Phillips SJ . Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA 2001; 285: 1719–28.
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 2004; 118: 687–98.
Chassagnon IR, McCarthy CA, Chin YK, Pineda SS, Keramidas A, Mobli M, et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc Natl Acad Sci U S A 2017; 114: 3750–5.
Pedersen SF, Klausen TK, Nilius B . The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol (Oxf) 2015; 213: 868–81.
Feustel PJ, Jin Y, Kimelberg HK . Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 2004; 35: 1164–8.
Alibrahim A, Zhao LY, Bae CY, Barszczyk A, Sun CL, Wang GL, et al. Neuroprotective effects of volume-regulated anion channel blocker DCPIB on neonatal hypoxic-ischemic injury. Acta Pharmacol Sin 2013; 34: 113–8.
Zhang Y, Zhang H, Feustel PJ, Kimelberg HK . DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol 2008; 210: 514–20.
Thompson RJ, Zhou N, MacVicar BA . Ischemia opens neuronal gap junction hemichannels. Science 2006; 312: 924–7.
Davidson JO, Drury PP, Green CR, Nicholson LF, Bennet L, Gunn AJ . Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS One 2014; 9: e96558.
Davidson JO, Green CR, Nicholson LF, O'Carroll SJ, Fraser M, Bennet L, et al. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann Neurol 2012; 71: 121–32.
Kozoriz MG, Bechberger JF, Bechberger GR, Suen MW, Moreno AP, Maass K, et al. The connexin43 C-terminal region mediates neuroprotection during stroke. J Neuropathol Exp Neurol 2010; 69: 196–206.
Fleig A, Penner R . The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci 2004; 25: 633–9.
Harteneck C . Function and pharmacology of TRPM cation channels. Naunyn Schmiedebergs Arch Pharmacol 2005; 371: 307–14.
Aarts MM, Tymianski M . TRPM7 and ischemic CNS injury. Neuroscientist 2005; 11: 116–23.
Bae CY, Sun HS . TRPM7 in cerebral ischemia and potential target for drug development in stroke. Acta Pharmacol Sin 2011; 32: 725–33.
Bae CY, Sun HS . Current understanding of TRPM7 pharmacology and drug development for stroke. Acta Pharmacol Sin 2013; 34: 10–6.
Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003; 115: 863–77.
Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci 2009; 12: 1300–7.
Coombes E, Jiang J, Chu XP, Inoue K, Seeds J, Branigan D, et al. Pathophysiologically relevant levels of hydrogen peroxide induce glutamate-independent neurodegeneration that involves activation of transient receptor potential melastatin 7 channels. Antioxid Redox Signal 2011; 14: 1815–27.
Jiang J, Li M, Yue L . Potentiation of TRPM7 inward currents by protons. J Gen Physiol 2005; 126: 137–50.
Clapham DE, Julius D, Montell C, Schultz G . International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev 2005; 57: 427–50.
Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, McNulty S . Tissue distribution profiles of the human TRPM cation channel family. J Recept Signal Transduct Res 2006; 26: 159–78.
Bai JZ, Lipski J . Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell death in organotypic hippocampal culture. Neurotoxicology 2010; 31: 204–14.
Chung KK, Freestone PS, Lipski J . Expression and functional properties of TRPM2 channels in dopaminergic neurons of the substantia nigra of the rat. J Neurophysiol 2011; 106: 2865–75.
Alim I, Teves L, Li R, Mori Y, Tymianski M . Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci 2013; 33: 17264–77.
Belrose JC, Xie YF, Gierszewski LJ, MacDonald JF, Jackson MF . Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol Brain 2012; 5: 11.
Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 2006; 101: 66–76.
Roedding AS, Tong SY, Au-Yeung W, Li PP, Warsh JJ . Chronic oxidative stress modulates TRPC3 and TRPM2 channel expression and function in rat primary cortical neurons: relevance to the pathophysiology of bipolar disorder. Brain Res 2013; 1517: 16–27.
Fonfria E, Mattei C, Hill K, Brown JT, Randall A, Benham CD, et al. TRPM2 is elevated in the tMCAO stroke model, transcriptionally regulated, and functionally expressed in C13 microglia. J Recept Signal Transduct Res 2006; 26: 179–98.
Jeong H, Kim YH, Lee Y, Jung SJ, Oh SB . TRPM2 contributes to LPC-induced intracellular Ca2+ influx and microglial activation. Biochem Biophys Res Commun 2017; 485: 301–6.
Mortadza SS, Sim JA, Stacey M, Jiang LH . Signalling mechanisms mediating Zn2+-induced TRPM2 channel activation and cell death in microglial cells. Sci Rep 2017; 7: 45032.
Miyake T, Shirakawa H, Kusano A, Sakimoto S, Konno M, Nakagawa T, et al. TRPM2 contributes to LPS/IFNgamma-induced production of nitric oxide via the p38/JNK pathway in microglia. Biochem Biophys Res Commun 2014; 444: 212–7.
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 2004; 286: C129–37.
Gelderblom M, Melzer N, Schattling B, Gob E, Hicking G, Arunachalam P, et al. Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke 2014; 45: 3395–402.
Alawieyah Syed Mortadza S, Sim JA, Neubrand VE, Jiang LH . A critical role of TRPM2 channel in Abeta42 -induced microglial activation and generation of tumor necrosis factor-alpha. Glia 2018; 66: 562–75.
Lee M, Cho T, Jantaratnotai N, Wang YT, McGeer E, McGeer PL . Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. FASEB J 2010; 24: 2533–45.
Di A, Kiya T, Gong H, Gao X, Malik AB . Role of the phagosomal redox-sensitive TRP channel TRPM2 in regulating bactericidal activity of macrophages. J Cell Sci 2017; 130: 735–44.
Zou J, Ainscough JF, Yang W, Sedo A, Yu SP, Mei ZZ, et al. A differential role of macrophage TRPM2 channels in Ca2+ signaling and cell death in early responses to H2O2 . Am J Physiol Cell Physiol 2013; 305: C61–9.
Lange I, Penner R, Fleig A, Beck A . Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium 2008; 44: 604–15.
Heiner I, Eisfeld J, Warnstedt M, Radukina N, Jungling E, Luckhoff A . Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J 2006; 398: 225–32.
Wang G, Cao L, Liu X, Sieracki NA, Di A, Wen X, et al. Oxidant sensing by TRPM2 inhibits neutrophil migration and mitigates inflammation. Dev Cell 2016; 38: 453–62.
Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, et al. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J 2011; 25: 3529–42.
Naziroglu M . TRPM2 channel membrane currents in primary rat megakaryocytes were activated by the agonist ADP-ribose but not oxidative stress. J Membr Biol 2011; 241: 51–7.
Hecquet CM, Ahmmed GU, Malik AB . TRPM2 channel regulates endothelial barrier function. Adv Exp Med Biol 2010; 661: 155–67.
Hecquet CM, Ahmmed GU, Vogel SM, Malik AB . Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res 2008; 102: 347–55.
Hecquet CM, Malik AB . Role of H2O2-activated TRPM2 calcium channel in oxidant-induced endothelial injury. Thromb Haemost 2009; 101: 619–25.
Hecquet CM, Zhang M, Mittal M, Vogel SM, Di A, Gao X, et al. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ Res 2014; 114: 469–79.
Mittal M, Nepal S, Tsukasaki Y, Hecquet CM, Soni D, Rehman J, et al. Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ Res 2017; 121: 1081–91.
Yang KT, Chang WL, Yang PC, Chien CL, Lai MS, Su MJ, et al. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ 2006; 13: 1815–26.
Ishii M, Shimizu S, Hara Y, Hagiwara T, Miyazaki A, Mori Y, et al. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium 2006; 39: 487–94.
Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, et al. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J 2006; 25: 1804–15.
Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R . TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci Signal 2009; 2: ra23.
McQuillin A, Bass NJ, Kalsi G, Lawrence J, Puri V, Choudhury K, et al. Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol Psychiatry 2006; 11: 134–42.
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 2002; 9: 163–73.
Hill K, Tigue NJ, Kelsell RE, Benham CD, McNulty S, Schaefer M, et al. Characterisation of recombinant rat TRPM2 and a TRPM2-like conductance in cultured rat striatal neurones. Neuropharmacology 2006; 50: 89–97.
Tong Q, Zhang W, Conrad K, Mostoller K, Cheung JY, Peterson BZ, et al. Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem 2006; 281: 9076–85.
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001; 411: 595–9.
Yu P, Xue X, Zhang J, Hu X, Wu Y, Jiang LH, et al. Identification of the ADPR binding pocket in the NUDT9 homology domain of TRPM2. J Gen Physiol 2017; 149: 219–35.
Fliegert R, Watt JM, Schobel A, Rozewitz MD, Moreau C, Kirchberger T, et al. Ligand-induced activation of human TRPM2 requires the terminal ribose of ADPR and involves Arg1433 and Tyr1349. Biochem J 2017; 474: 2159–75.
Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem 2005; 280: 6138–48.
Perraud AL, Schmitz C, Scharenberg AM . TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium 2003; 33: 519–31.
Mei ZZ, Xia R, Beech DJ, Jiang LH . Intracellular coiled-coil domain engaged in subunit interaction and assembly of melastatin-related transient receptor potential channel 2. J Biol Chem 2006; 281: 38748–56.
McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ . Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem 2003; 278: 11002–6.
Starkus J, Beck A, Fleig A, Penner R . Regulation of TRPM2 by extra- and intracellular calcium. J Gen Physiol 2007; 130: 427–40.
Du J, Xie J, Yue L . Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci U S A 2009; 106: 7239–44.
Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A . Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem 2002; 277: 23150–6.
Uemura T, Kudoh J, Noda S, Kanba S, Shimizu N . Characterization of human and mouse TRPM2 genes: identification of a novel N-terminal truncated protein specifically expressed in human striatum. Biochem Biophys Res Commun 2005; 328: 1232–43.
Orfanelli U, Wenke AK, Doglioni C, Russo V, Bosserhoff AK, Lavorgna G . Identification of novel sense and antisense transcription at the TRPM2 locus in cancer. Cell Res 2008; 18: 1128–40.
Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, et al. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 2001; 293: 1327–30.
Inamura K, Sano Y, Mochizuki S, Yokoi H, Miyake A, Nozawa K, et al. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol 2003; 191: 201–7.
Xia R, Mei ZZ, Mao HJ, Yang W, Dong L, Bradley H, et al. Identification of pore residues engaged in determining divalent cationic permeation in transient receptor potential melastatin subtype channel 2. J Biol Chem 2008; 283: 27426–32.
Mei ZZ, Mao HJ, Jiang LH . Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am J Physiol Cell Physiol 2006; 291: C1022–8.
Hermosura MC, Cui AM, Go RC, Davenport B, Shetler CM, Heizer JW, et al. Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc Natl Acad Sci U S A 2008; 105: 18029–34.
Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol 2004; 143: 186–92.
Fonfria E, Marshall IC, Boyfield I, Skaper SD, Hughes JP, Owen DE, et al. Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem 2005; 95: 715–23.
Pang B, Shin DH, Park KS, Huh YJ, Woo J, Zhang YH, et al. Differential pathways for calcium influx activated by concanavalin A and CD3 stimulation in Jurkat T cells. Pflugers Arch 2012; 463: 309–18.
Roberge S, Roussel J, Andersson DC, Meli AC, Vidal B, Blandel F, et al. TNF-alpha-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc Res 2014; 103: 90–9.
Kolisek M, Beck A, Fleig A, Penner R . Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 2005; 18: 61–9.
Grubisha O, Rafty LA, Takanishi CL, Xu X, Tong L, Perraud AL, et al. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J Biol Chem 2006; 281: 14057–65.
Fliegert R, Gasser A, Guse AH . Regulation of calcium signalling by adenine-based second messengers. Biochem Soc Trans 2007; 35: 109–14.
Toth B, Iordanov I, Csanady L . Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J Gen Physiol 2015; 145: 419–30.
Beck A, Kolisek M, Bagley LA, Fleig A, Penner R . Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J 2006; 20: 962–4.
Du J, Xie J, Yue L . Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J Gen Physiol 2009; 134: 471–88.
Starkus JG, Fleig A, Penner R . The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J Physiol 2010; 588: 1227–40.
Yang W, Zou J, Xia R, Vaal ML, Seymour VA, Luo J, et al. State-dependent inhibition of TRPM2 channel by acidic pH. J Biol Chem 2010; 285: 30411–8.
Zeng B, Chen GL, Xu SZ . Divalent copper is a potent extracellular blocker for TRPM2 channel. Biochem Biophys Res Commun 2012; 424: 279–84.
Yang W, Manna PT, Zou J, Luo J, Beech DJ, Sivaprasadarao A, et al. Zinc inactivates melastatin transient receptor potential 2 channels via the outer pore. J Biol Chem 2011; 286: 23789–98.
Yu W, Jiang LH, Zheng Y, Hu X, Luo J, Yang W . Inactivation of TRPM2 channels by extracellular divalent copper. PLoS One 2014; 9: e112071.
Naziroglu M, Luckhoff A, Jungling E . Antagonist effect of flufenamic acid on TRPM2 cation channels activated by hydrogen peroxide. Cell Biochem Funct 2007; 25: 383–7.
Hill K, McNulty S, Randall AD . Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch Pharmacol 2004; 370: 227–37.
Togashi K, Inada H, Tominaga M . Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol 2008; 153: 1324–30.
Kraft R, Grimm C, Frenzel H, Harteneck C . Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol 2006; 148: 264–73.
Shimizu S, Yonezawa R, Hagiwara T, Yoshida T, Takahashi N, Hamano S, et al. Inhibitory effects of AG490 on H2O2-induced TRPM2-mediated Ca2+ entry. Eur J Pharmacol 2014; 742: 22–30.
Yamamoto S, Toda T, Yonezawa R, Negoro T, Shimizu S . Tyrphostin AG-related compounds attenuate H2O2-induced TRPM2-dependent and -independent cellular responses. J Pharmacol Sci 2017; 134: 68–74.
Starkus JG, Poerzgen P, Layugan K, Kawabata KG, Goto JI, Suzuki S, et al. Scalaradial is a potent inhibitor of transient receptor potential melastatin 2 (TRPM2) ion channels. J Nat Prod 2017; 80: 2741–50.
Zhang W, Tong Q, Conrad K, Wozney J, Cheung JY, Miller BA . Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am J Physiol Cell Physiol 2007; 292: C1746–58.
Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, et al. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol 2009; 587: 965–79.
Ye M, Yang W, Ainscough JF, Hu XP, Li X, Sedo A, et al. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis 2014; 5: e1541.
Huang S, Turlova E, Li F, Bao MH, Szeto V, Wong R, et al. Transient receptor potential melastatin 2 channels (TRPM2) mediate neonatal hypoxic-ischemic brain injury in mice. Exp Neurol 2017; 296: 32–40.
Huang S, Wang H, Turlova E, Abussaud A, Ji X, Britto LR, et al. GSK-3beta inhibitor TDZD-8 reduces neonatal hypoxic-ischemic brain injury in mice. CNS Neurosci Ther 2017; 23: 405–15.
Jia J, Verma S, Nakayama S, Quillinan N, Grafe MR, Hurn PD, et al. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J Cereb Blood Flow Metab 2011; 31: 2160–8.
Verma S, Quillinan N, Yang YF, Nakayama S, Cheng J, Kelley MH, et al. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci Lett 2012; 530: 41–6.
Nakayama S, Vest R, Traystman RJ, Herson PS . Sexually dimorphic response of TRPM2 inhibition following cardiac arrest-induced global cerebral ischemia in mice. J Mol Neurosci 2013; 51: 92–8.
Shimizu T, Dietz RM, Cruz-Torres I, Strnad F, Garske AK, Moreno M, et al. Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp Neurol 2016; 283: 151–6.
Shimizu T, Macey TA, Quillinan N, Klawitter J, Perraud AL, Traystman RJ, et al. Androgen and PARP-1 regulation of TRPM2 channels after ischemic injury. J Cereb Blood Flow Metab 2013; 33: 1549–55.
Quillinan N, Grewal H, Klawitter J, Herson PS . Sex steroids do not modulate TRPM2-mediated injury in females following middle cerebral artery occlusion(1,2,3). eNeuro 2014; 1. pii: ENEURO.0022-14.2014.
Patel AR, Ritzel R, McCullough LD, Liu F . Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol 2013; 5: 73–90.
Aminzadeh M, Roghani M, Sarfallah A, Riazi GH . TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer's disease. Int Immunopharmacol 2017; 54: 78–85.
Tseng HH, Vong CT, Kwan YW, Lee SM, Hoi MP . TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci Rep 2016; 6: 35016.
Sofroniew MV . Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 2009; 32: 638–47.
Endoh M, Maiese K, Wagner J . Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res 1994; 651: 92–100.
McKeon RJ, Schreiber RC, Rudge JS, Silver J . Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci 1991; 11: 3398–411.
Wang J, Jackson MF, Xie YF . Glia and TRPM2 channels in plasticity of central nervous system and Alzheimer's diseases. Neural Plast 2016; 2016: 1680905.
Sandoval KE, Witt KA . Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008; 32: 200–19.
Brown RC, Wu L, Hicks K, O'neil RG . Regulation of blood-brain barrier permeability by transient receptor potential type C and type v calcium-permeable channels. Microcirculation 2008; 15: 359–71.
Jiang Q, Gao Y, Wang C, Tao R, Wu Y, Zhan K, et al. Nitration of TRPM2 as a molecular switch induces autophagy during brain pericyte injury. Antioxid Redox Signal 2017; 27: 1297–316.
Zhang Y, Zhang X, Park TS, Gidday JM . Cerebral endothelial cell apoptosis after ischemia-reperfusion: role of PARP activation and AIF translocation. J Cereb Blood Flow Metab 2005; 25: 868–77.
Buelow B, Song Y, Scharenberg AM . The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem 2008; 283: 24571–83.
Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, et al. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 2008; 112: 2770–9.
Herz J, Sabellek P, Lane TE, Gunzer M, Hermann DM, Doeppner TR . Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke 2015; 46: 2916–25.
Acknowledgements
This work was supported by the following grants: Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants to Zhong-Ping FENG (RGPIN-2014-06471) and to Hong-Shuo SUN (RGPIN-2016-04574), and a NSERC Alexander Graham Bell Canada Graduate Scholarship to Ekaterina TURLOVA (CGS-D).
Author information
Authors and Affiliations
Corresponding authors
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Turlova, E., Feng, Zp. & Sun, Hs. The role of TRPM2 channels in neurons, glial cells and the blood-brain barrier in cerebral ischemia and hypoxia. Acta Pharmacol Sin 39, 713–721 (2018). https://doi.org/10.1038/aps.2017.194
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.194
Keywords
This article is cited by
-
Pathological Mechanisms Induced by TRPM2 Ion Channels Activation in Renal Ischemia-Reperfusion Injury
Molecular Biology Reports (2022)
-
Umbilical cord-derived mesenchymal stem cell conditioned medium reverses neuronal oxidative injury by inhibition of TRPM2 activation and the JNK signaling pathway
Molecular Biology Reports (2022)
-
Paraquat as an Environmental Risk Factor in Parkinson’s Disease Accelerates Age-Related Degeneration Via Rapid Influx of Extracellular Zn2+ into Nigral Dopaminergic Neurons
Molecular Neurobiology (2019)
-
A new platform for international collaboration on pharmacology and drug development: 2017 China-Canada-USA Pharmacology/Physiology Conference
Acta Pharmacologica Sinica (2018)
