
Abstract
Non-alcoholic fatty liver disease (NAFLD) is a clinical syndrome characterized by hepatic steatosis. NAFLD is closely linked to obesity, insulin resistance and dyslipidemia. AMP-activated protein kinase (AMPK) functions as an energy sensor and plays a central role in regulating lipid metabolism. In this study, we identified a series of novel pyrazolone AMPK activators using a homogeneous time-resolved fluorescence assay (HTRF) based on the AMPKα2β1γ1 complex. Compound 29 (C29) is a candidate compound that directly activated the kinase domain of AMPK with an EC50 value of 2.1–0.2 μmol/L and acted as a non-selective activator of AMPK complexes. Treatment of HepG2 cells with C29 (20, 40 μmol/L) dose-dependently inhibited triglyceride accumulation. Chronic administration of C29 (10, 30 mg/kg every day, po, for 5 weeks) significantly improved lipid metabolism in both the liver and the plasma of ob/ob mice. These results demonstrate that the AMPK activators could be part of a novel treatment approach for NAFLD and associated metabolic disorders.
Similar content being viewed by others

Introduction
Nonalcoholic fatty liver disease (NAFLD) is found in 20%−30% of the general population worldwide and represents a spectrum of hepatic dysfunction linked to obesity, insulin resistance and metabolic syndrome1,2,3,4. NAFLD exhibits dysregulation of hepatic lipid metabolism and accelerates the accumulation of triglycerides and cholesterol in hepatocytes5. The excess accumulation of lipids predisposes the liver to additional proinflammatory conditions and together with the dysfunction of adipose tissue exacerbates hepatic steatosis6,7. Hepatic steatosis is a leading cause of nonalcoholic steatohepatitis (NASH) and can cause fibrosis, cirrhosis and hepatocellular carcinoma (HCC). Multiple important pathogenic drivers have been implicated in the initiation and progression of hepatic steatosis, such as lipotoxicity, oxidative stress and immune cell infiltration8,9. However, there is no well-established pharmacological approach for the treatment of hepatic steatosis10,11. Thus, studies identifying novel therapeutic targets and NAFLD treatments are urgently needed.
AMP-activated protein kinase (AMPK) is a conserved serine/threonine protein kinase that is activated by low cellular energy status. The activation of AMPK restores cellular energy homeostasis by triggering catalytic processes (eg, glucose and fatty acid oxidation) to stimulate ATP generation and inhibits anabolic processes (eg, gluconeogenesis and fatty acid synthesis) to decrease ATP consumption12. Due to its central role in the control of multiple metabolic pathways, AMPK has become a potential therapeutic target for treating type 2 diabetes (T2D), insulin resistance, obesity and NAFLD13,14,15.
AMPK exists as a heterotrimer consisting of a highly conserved catalytic α subunit and regulatory β and γ subunits. In mammals, each subunit is encoded by several distinct genes (α1, α2; β1, β2; γ1, γ2 and γ3), and there are at least 12 possible distinct AMPKαβγ heterotrimeric complexes16. There is differential expression of the complexes, and the expression profile is unique in specific tissues or subcellular locations. The α subunit is composed of a kinase domain (KD), an autoinhibitory domain (AID), and a C-terminal β subunit-binding domain. The β subunit is composed of a glycogen-binding domain and a C-terminal α and γ subunit-binding domain. The γ subunit has a β subunit-binding region and four tandem repeats of cystathionine-β-synthase (CBS) motifs12. The mammalian AMPK heterotrimers are activated in the following three complementary ways: (1) phosphorylation of the conserved threonine residue within the KD (Thr172 in rat α2; the number may be different in other species) by upstream kinases (tumor-suppressor liver kinase B1, calcium/calmodulin-dependent protein kinase kinases and transforming growth factor-β activated protein kinase-1), which causes a >100-fold increase in kinase activity; (2) inhibition of Thr172 dephosphorylation by protein phosphatase 2Cα (PP2Cα); and (3) allosteric activation by AMP binding to the CBS domains in the γ subunit, which leads to conformational changes and induces Thr172 phosphorylation by upstream kinases and concurrent inhibition of Thr172 dephosphorylation17,18,19.
Numerous observations obtained using agents such as metformin and berberine to deplete intracellular ATP or a pharmacological activator indicated that AMPK activation decreases the hepatic triglyceride (TG) content by decreasing de novo lipogenesis, promoting fatty acid oxidation and regulating inflammation20,21,22. Additionally, in a genetic mouse model, AMPK activation specific to the liver protected against hepatic steatosis when the mice were fed a high fructose diet, and the AMPK β1/β2 knockout specific to the adipose tissue increased HFD-induced hepatic lipid accumulation23,24. These findings confirm the hypothesis that pharmacological activation of AMPK could provide a new strategy for the management of NAFLD.
Given the potential therapeutic application of AMPK activators, the feasibility of developing AMPK activators has begun to emerge25. A769662, the first AMPK direct activator identified by Abbott laboratories in 2006, binds to the allosteric site between the α and β subunits of AMPK20,25,26. Several structurally diverse compounds such as 991, PF-06409577 and PF-249 have also been shown to bind to this allosteric pocket27,28,29. In addition, a series of AMPK activators with different binding sites such as AICAR (ZMP), C13 (C2), MT63-78, GSK621, PF-739, PF-249, and MK-8722 have been identified by different laboratories28,30,31,32,33,34,35. In our previous work, we identified a new class of AMPK activators by high-throughput screening. These molecules activate AMPK by antagonizing the auto-inhibition of the α subunit of AMPK36,37,38.
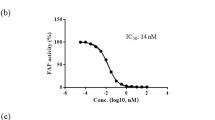
More recently, we identified a novel AMPK activator, compound 1 (Figure 1), using an HTRF assay in vitro (Figure S1). Compound 1 activates the purified human AMPKα2β1γ1 complex (EC50=5.2 μmol/L). However, the compound showed poor plasma stability in vivo. Herein, we present our efforts to optimize compound 1 that led to the identification of substituted pyrazolone derivatives as novel AMPK activators.

Structure of compound 1.
Materials and methods
Chemistry
The experimental procedures and characterization of all compounds are provided in the Supporting Information.
Different recombinant AMPK protein construction, expression and purification
The coding sequences of the α/β/γ subunits of various AMPK heterotrimers were amplified from the cDNA of each human AMPK subunit and constructed in a pET28b vector, the different AMPK α subunit truncations were constructed in the pET28b vector and expressed and purified as previously described39,40.
Measurement of AMPK activity in vitro
The indicated AMPK heterotrimers and α subunit truncations were fully phosphorylated by CaMKKβ as previously described. In the HTRF assay, AMPK activity and the activation of compounds by AMPK were detected using the STK substrate 1-biotin, XL-665 and the STK-antibody of HTRF® KinEASE™-STK1 Kit as previously described41. The activation fold change was compared to the baseline activity of AMPK completely phosphorylated by CaMKKβ. For the filter assay, the AMPK activity and the activation of compounds by AMPK were detected mainly as previously described except the reactions were conducted in 96-well V-shape plates with a 50 μL mixture containing 32 mmol/L Tris-HCl, pH 7.5, 4 mmol/L MgCl2, 0.8 mmol/L DTT, 2% DMSO (compounds were dissolved in DMSO), 2 μmol/L SAMS peptide and 10 μmol/L ATP (0.2 μCi of [γ-33P] ATP per reaction). The reaction was initiated by the addition of AMPKα2β1γ1 (final concentration 2.5 nmol/L), incubated at 30 °C for 45 min, and terminated by the addition of 50 μL 200 mmol/L H3PO4. We then transferred 40 μL of mixture to phosphocellulose filter plates (Millipore) that were pre-wet with 1 mmol/L Tris-HCl, pH 7.5. The plates were washed 3 times using 200 mmol/L H3PO4. The final step was to add 150 μL liquid scintillation counting cocktail (PerkinElmer) into each well before detecting the radioactivity on a Wallac Microbeta plate counter37.
Measurement of lipid synthesis in the HepG2 cell line
A total of 4×104 HepG2 cells were seeded per well in white-walled 96-well plates containing HG-DMEM supplemented with 10% FBS. After 24 h of incubation, the cells were deprived of FBS for 2 h, and followed by 20 h of C29 incubation with serum-free HG-DMEM. Then, 20 μL of serum-free HG-DMEM containing 14C acetic sodium (0.1 μCi/mL, Perkin Elmer) was added to each well and incubated for 4 h. The plates were rinsed with cold PBS, and the final wash was replaced with 0.25 mol/L NaOH. The protein concentration was then measured, and Microscint 20 was added to wells. The radioactivity incorporated into the lipids was monitored using a Wallac Microbeta plate reader and corrected for protein concentration42.
Adenovirus infection and measurement of lipid synthesis
The dominant negative forms of AMPK, AMPKα1 (D159A) and AMPKα2 (K45R) (α1/α2-DN), were constructed by using the pAdEasy system (Agilent Technologies, New York, CA, USA). We seeded 4×104 HepG2 cells per well in white-walled 96-well plates with HG-DMEM supplemented with 10% FBS. After 12 h, the HepG2 cells were infected with adenovirus expressing control GFP or α1/α2-DN for 6 h before the medium was replaced with HG-DMEM supplemented with 10% FBS. The cells were cultured for 2 days before the experiments were initiated.
L6 myotube culture and 2-deoxy-[3H]-D-glucose uptake measurements
We seeded 4×105 myoblast cells per well in 24-well plates containing HG-DMEM with 10% FBS medium, and when the cells reached 90% confluence, the medium was replaced with differentiation medium (HG-DMEM containing 2% FBS). Four days after differentiation, the L6 myotube cells were used for compound treatment and subsequent glucose uptake measurements as described previously. Briefly, after stimulation with C29, the cells were washed three times with HBSS buffer, followed by incubation with 2-deoxy-3H]-D-glucose for 15 min. The cells were washed with ice-cold PBS and lysed using 0.25 mol/L NaOH before the radioactivity was counted43.
Statistical analysis
The results are presented as the mean±SEM. Differences between the two groups were analyzed using paired Student's t-tests. The differences among multiple groups were compared by one-way ANOVA, followed by LSD comparison. P<0.05 was regarded as significant.
Results
Structure and activity relationship of compound 1 derivatives
Due to the instability of the ester group in compound 1, we initially prepared compounds 16-25 with either an alkyl amine (16-20, 23, 25) or an arylamine (21, 22, 24) instead of an ester group. The activities of these compounds are summarized in Table 1. Among these pyrazolones, compounds 16 and 21-24 showed no AMPK activation at 20 μmol/L. However, the other compounds showed a range of AMPK activation activities at 20 μmol/L. No arylamine-substituted pyrazolones showed any activity (21, 22 and 24), but the alkylamine pyralozones remained active. The trends showed that larger substituents significantly affected the activity (17, 18, 19). In particular, compounds with a phenyl-substituted ring showed better activity. Moreover, an electron-donating group on the phenyl ring (25) enhanced activity, and an electron withdrawing substituent reduced activity (23).
Based on these preliminary structure–activity relationships (SARs), a follow-up-focused library that systematically varied the right-hand side benzene ring subunit of compound 1 was synthesized. We chose electron-donating groups from the library for the phenyl ring compound. The activities of these compounds toward AMPK are summarized in Table 2. Most of this series of compounds showed potent activity. Among the 12 compounds tested (26–37), 8 were more active than the lead compound 1. In particular, 3,4-dimethoxy-substituted compound 29 showed the best activity (EC50=2.1 μmol/L, fold=3.3). The overall SAR indicates that the disubstituted compounds are more active than the mono-substituted and trisubstituted compounds (compounds 29, 25, 30).
After synthesizing the potent activator compound 29, we then addressed the left-hand side modifications in an attempt to further improve potency. The 3,4-dimethoxy on the right side of the benzene ring was maintained, and we investigated the groups on the left side. Compounds 38–45 (Table 3) were then synthesized. Except for compound 41, with a 3-trifluoromethyl group on the left-hand benzene ring and an activity (EC50=5.6 μmol/L, fold=3.2) similar to that of compound 29 (EC50=2.1 μmol/L, fold=3.3), the other compounds exhibited decreased activity compared to compound 29.
We next studied the middle of the structure by keeping the left and right favored substituent groups, including 4-fluoro-3-trifluoromethyl phenyl and 3,4-dimethoxy benzyl. Compounds 46 and 47 were designed to determine the skeleton of the piperidine pyrazolone, while 48 and 49 were designed to determine substituents and sites in the middle benzene ring relative to its activity (Table 4). Compound 47 had a methyl group on the pyrazolone ring N atom, and its activity (EC50=3.3 μmol/L, fold=3.6) was retained. The other three compounds also generally retained their activity.
In in vitro metabolism studies of compound 29 in human, monkey, dog, mouse and rat liver microsomes, compound 29 was metabolized by 48.7% after incubation for 60 min in human liver microsomes and may be metabolized through cleavage of the amide bond. The results of liver microsomal metabolism also indicated that monkey and mouse have similar metabolic characteristics as humans, preventing amide bond cleavage and increasing the stability of compound 29. Compounds 50–54 were synthesized (Table 5), and the N atom of the amide bond was alkylated with groups of various sizes (52, 53) or α-benzyl substituted (50, 51) or the distance between the phenyl and the N atom was increased (54). Compound 52, with a methyl group on the amide N atom, showed improved activity (EC50=1.1 μmol/L, fold=3.3) compared to compound 29. However, compounds 52 and 54 showed lower stability than compound 29 in an in vitro liver microsome assay.
C29 directly activates AMPK heterotrimers and the kinase domain
After observation of the activation of AMPK in the in vitro HTRF assay and metabolic stability improvement in liver microsomes, compound 29 (C29) was selected for further mechanistic exploration. First, we wondered whether C29 could activate AMPK heterotrimers consisting of different α/β/γ subunits. As reported44, A769662 could only activate AMPK heterotrimers containing the β1 subunit, but C29 activated different AMPK heterotrimers with similar EC50 values (Figure 2A, 2B and Table S1). These results indicate that in contrast to A769662, C29 was a non-β subunit-selective AMPK activator. The filter assay results also showed that C29 could activate AMPKα2β1γ1 directly (Figure 2C, 2D and Table S1). C29 simultaneously activated both α subunit truncations, with an EC50 of 3.6 μmol/L and 4.0 μmol/L for α1 (1-394) and α2 (1-398) respectively (Figure 3A). These results are similar to the heterotrimer data and suggest that unlike AMP, C29 activated AMPK independently of the γ subunit. To further address this issue, we investigated how C29 activated AMPKα2β1γ1 in a dose-responsive manner in the presence of 50 nmol/L A769662 or 10 μmol/L AMP, which are saturating concentrations for these molecules (Figure 3B, 3C). Both A769662 and AMP activated AMPK dose-dependently in the presence of a maximally efficacious concentration of C29 (Figure 3D, 3E). Interestingly, we tested C29 on α subunit truncations containing only the kinase domain: α1(11-281) and α2(1-285). The results showed that C29 activated α1 (11-281) and α2 (1-285) in a dose-dependent manner, with EC50 values of 3.7 μmol/L and 6.8 μmol/L, respectively (Figure 3F). These results indicated that C29 directly activated the AMPK kinase domain.

C29 activated AMPK heterotrimers without subunit selectivity. (A) A769662-activated AMPK heterotrimers containing β1 subunit. (B) C29-activated AMPK heterotrimers with different subunit complexes. (C,D) The AMPK activation curve of A769662, and C29 on AMPKα2β1γ1 in Filter assay. The results are shown as the mean±SEM.

C29 activated AMPK kinase domain directly. (A) C29-activated AMPK α subunit containing autoinhibitory domain, α1(1-394) and α2(1-398). (B,C) Additive activation of AMPKα2β1γ1 by different concentrations of C29 with 50 nmol/L A769662 (B) or 10 μmol/L AMP (C). (D, E) Additive activation of AMPKα2β1γ1 with different concentrations of A769662 (D) or AMP (E) with 40 μmol/L C29. (F) C29 activated AMPK kinase domain, α1(11-281) and α2(1-285). The results are shown as the mean±SEM.
C29 activated AMPK and inhibited triglyceride accumulation in hepatocytes
Activation of AMPK regulates various downstream target proteins including acetyl coenzyme A carboxylase (ACC1 and ACC2), which play important role in controlling fatty acid metabolism. After we observed the allosteric activation of AMPK on a molecular level, we next investigated the effect of C29 in hepatocytes. The results showed that C29 activated AMPK and stimulated phosphorylation of ACC dose-dependently in rat primary hepatocytes and the HepG2 hepatocellular carcinoma cell line after 1 h of treatment (Figure 4A, 4B). The treatment had no effect on the ratio of AMP/ATP and ADP/ATP in primary hepatocytes (Figure 4C). The activation of AMPK decreases the lipid content in hepatocytes through inhibition of de novo lipogenesis (DNL) and by increasing fatty acid oxidation by stimulation of phosphorylation of ACC1 and ACC2. As expected, C29 decreased the expression of SREBP-1c (Figure S2) and inhibited the accumulation of triglycerides in a dose-dependent manner in HepG2 cells (Figure 4D). Additionally, both the non-specific AMPK inhibitor compound C and the AMPK DN (dead kinase) reversed intracellular triglyceride accumulation after C29 treatment (Figure 4E, 4F). These results suggest that C29 activated AMPK and inhibited accumulation of triglycerides likely via an AMPK-dependent pathway in hepatocytes. We also found that C29 could activate AMPK does-dependently in L6 myotube cells without increasing the AMP/ATP and ADP/ATP ratio and stimulated glucose uptake in an AMPK-dependent manner (Figure S3).

C29 improved lipid metabolism in an AMPK signaling pathway-dependent manner. (A,B) C29 dose-dependently activated AMPK in the primary hepatocytes (A) and the HepG2 cell line (B) after 1 h of incubation; A769662 (40 μmol/L) was used as a positive control. (C) C29 did not change the AMP/ATP and ADP/ATP ratio after a 3-h incubation in rat primary hepatocytes. CCCP (10 μmol/L) was used as a positive control. (D) C29 inhibited triglyceride accumulation in a dose-dependent manner in HepG2 cells after a 24-h incubation. (E, F) Compound C and AMPK DN blocked triglyceride accumulation inhibition induced by C29 (20 μmol/L). The results are shown as the mean±SEM. * P<0.05, ** P<0.01.
Oral efficacy of C29 reduced lipid accumulation in ob/ob mice
We then tested C29 using in vivo studies due to the observed beneficial cellular effects in hepatocytes and used metformin as the positive control. Following oral administration at a concentration of 10 mg/kg, C29 was rapidly absorbed (T max=0.5 h) (Figure S4). The desirable in vivo PK profile prompted us to further evaluate its effects on metabolic syndrome in ob/ob mice. The compound was administered for 5 weeks, and we found that C29 lowered ob/ob mouse body weight and decreased both liver weight and abdominal fat (Figure 5A–5C) while simultaneously decreasing plasma triglycerides (Figure 5D). The reduced liver weight was inferred from the reduction of hepatic triglyceride and cholesterol accumulation (Figure 5E, 5F) and a significant increase in the AMPK signaling pathway in the liver (Figure 6). These results support the hypothesis that the AMPK activator C29 can potentially reduce lipid accumulation in the liver and plasma in ob/ob mice and may provide therapeutic benefits for NAFLD patients.

Treatment of C29 improved lipid metabolism in the liver of ob/ob mice. (A) Body weight of the mice after treatment by vehicle (Veh), positive control metformin of 250 mg/kg (Met), C29 of 10 or 30 mg/kg (n=8–10). (B, C) C29 decreased the liver and fat mass of ob/ob mice after treatment. (D) C29 decreased the triglyceride content in the plasma. (E, F) C29 decreased hepatic triglyceride and cholesterol accumulation. The results are shown as the mean±SEM. * P<0.05, ** P<0.01 vs Veh.

C29 increased hepatic AMPK activity in ob/ob mice after a 5-week treatment. The ratio of phosphorylation level to protein level of AMPK and ACC was determined. The results are shown as the mean±SEM. * P<0.05, ** P<0.01.
Discussion
AMPK, an energy sensor involved in a combination of inhibited anabolic pathways and stimulated catabolic pathways, has been identified as a potential drug target for metabolic diseases such as diabetes and NAFLD12,22,24,44. Current AMPK allosteric activators could be divided into three classes: activators bind to the CBS-motif on γ subunit of AMPK such as AMP and AICAR; activators interact with the ADaM domain such as A769662 and 991; and the third class of activators such as PT1 activate AMPK by antagonizing the autoinhibition of the AMPKα subunit20,29,34,37. Here, we report a novel AMPK activator named C29 identified with our HTRF assay. C29 activated the AMPKα kinase domain directly (Figure 3A, 3F) and allowed it to activate AMPK heterotrimers with non-selective properties (Figure 2A, 2B). This approach may be a new mechanism for AMPK allosteric activation, and more work to identify the binding site will be required for us to gain insights into the molecular basis of the pan-AMPK activity of C29.
AMPK activation in hepatocytes involves a combination of inhibition of lipogenesis and stimulation of fatty acid oxidation, which is an attractive profile for NAFLD patients. In this study, we showed that C29 treatment activated AMPK dose dependently (Figure 4A, 4B) and leads to a dramatic reduction in triglyceride accumulation in hepatocytes (Figure 4D–4F). We found that C29 treatment in vivo potentially increased phosphorylation of hepatic AMPK after a 5-week dosing period (Figure 6). The decrease in plasma and liver lipid levels can be explained by the decreased synthesis of fatty acids in the liver (Figure 5D–5F).
In contrast to activation in the liver, activation of AMPK in the skeletal muscle is associated with glucose consumption. MK-8722 and PF739 are two systemic pan-AMPK activators that mediate AMPK activation in skeletal muscle and induce glucose uptake, which lowers glucose levels in rodents and non-human primates28,32. Despite maintaining an obvious glucose uptake in L6 myotubes in an AMPK-dependent pathway (Figure S3), C29 exhibited a slight ability to improve glucose tolerance in ob/ob mice after chronic dosing. This effect may be consistent with the poor distribution and weak activation of AMPK in the muscle after oral treatment with C29 (data not shown). Additional studies are needed to confirm the efficacy of C29 for glucose lowering in vivo. Besides, previous studies reported that chronic systemic AMPK activation could induce cardiac hypertrophy in a cardiac glycogen-dependent or glycogen-independent mechanism32,45. We found no adverse effects (including the activity of creatine phosphokinase) after oral doses of C29 at 300 mg/kg for 14 d in mice, which indicates C29 does not impact cardiac function (data not shown).
In summary, we report a novel series of pyrazolone derivatives as direct non-selective activators of AMPK heterotrimers and the kinase domain. After initial structural modification of the right ester group to an amide and optimization of the left side and the linking part of compound 1 to improve its potency and in vitro stability, C29 was selected as a typical compound that stimulated phosphorylation of AMPK and ACC without affecting the AMP/ATP and ADP/ATP ratios. C29 also inhibited lipid accumulation in hepatocytes and improved lipid metabolism both in the liver and in the plasma of ob/ob mice. These results indicate that C29 is a novel allosteric AMPK activator and has potential applications in the treatment of NAFLD and associated metabolic disorders.
Author contribution
Mei ZHANG and Zhi-fu XIE conducted the key experiments, contributed equally to this work. Mei ZHANG, Zhi-fu XIE, Da-kai CHEN, Jia LI, Fa-jun NAN, and Jing-ya LI conceived and designed the studies, analyzed the initial data and wrote the manuscript. Mei ZHANG, Run-tao ZHANG, and Yang-ming ZHANG conducted the structure-activity relationship analysis, Min GU performed the HPLC analysis. Shi-chao CUI, Yan-yan YU, and Xin-wen ZHANG performed some of the cell and animal studies.
References
Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol 2016; 78: 181–205.
Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol 2016; 11: 451–96.
Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013; 10: 686–90.
Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413–9.
Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 2008; 118: 829–38.
Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002; 346: 1221–31.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012; 482: 179–85.
Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016; 13: 88–110.
Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010; 52: 774–88.
Brodosi L, Marchignoli F, Petroni ML, Marchesini G. NASH: a glance at the landscape of pharmacological treatment. Ann Hepatol 2016; 15: 673–81.
Cassidy S, Syed BA. Nonalcoholic steatohepatitis (NASH) drugs market. Nat Rev Drug Discov 2016; 15: 745–6.
Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 2016; 26: 190–201.
Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol 2017; 45: 31–7.
Day EA, Ford RJ, Steinberg GR. AMPK as a therapeutic target for treating metabolic diseases. Trends Endocrinol Metab 2017; 28: 545–60.
Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005; 1: 15–25.
Quentin T, Kitz J, Steinmetz M, Poppe A, Bar K, Kratzner R. Different expression of the catalytic alpha subunits of the AMP activated protein kinase–an immunohistochemical study in human tissue. Histol Histopathol 2011; 26: 589–96.
Davies SP, Helps NR, Cohen PT, Hardie DG. 5'-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 1995; 377: 421–5.
Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 2013; 18: 556–66.
Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 2007; 403: 139–48.
Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006; 3: 403–16.
Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 2013; 19: 1649–54.
Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab 2016; 311: E730–E40.
Mottillo EP, Desjardins EM, Crane JD, Smith BK, Green AE, Ducommun S, et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab 2016; 24: 118–29.
Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly YM, et al. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep 2017; 18: 3043–51.
Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem 2007; 282: 32539–48.
Calabrese MF, Rajamohan F, Harris MS, Caspers NL, Magyar R, Withka JM, et al. Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 2014; 22: 1161–72.
Cameron KO, Kung DW, Kalgutkar AS, Kurumbail RG, Miller R, Salatto CT, et al. Discovery and preclinical characterization of 6-chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic acid (pf-06409577), a direct activator of adenosine monophosphate-activated protein kinase (AMPK), for the potential treatment of diabetic nephropathy. J Med Chem 2016; 59: 8068–81.
Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjobsted R, Jorgensen NO, et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab 2017; 25: 1147–59 e10.
Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun 2013; 4: 3017.
Gomez-Galeno JE, Dang Q, Nguyen TH, Boyer SH, Grote MP, Sun Z, et al. A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med Chem Lett 2010; 1: 478–82.
Hunter RW, Foretz M, Bultot L, Fullerton MD, Deak M, Ross FA, et al. Mechanism of action of compound-13: an alpha1-selective small molecule activator of AMPK. Chem Biol 2014; 21: 866–79.
Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017; 357: 507–11.
Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, et al. Co-activation of AMPK and mTORC1 induces cytotoxicity in acute myeloid leukemia. Cell Rep 2015; 11: 1446–57.
Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett 1994; 353: 33–6.
Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med 2014; 6: 519–38.
Li YY, Yu LF, Zhang LN, Qiu BY, Su MB, Wu F, et al. Novel small-molecule AMPK activator orally exerts beneficial effects on diabetic db/db mice. Toxicol Appl Pharmacol 2013; 273: 325–34.
Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem 2008; 283: 16051–60.
Yu LF, Li YY, Su MB, Zhang M, Zhang W, Zhang LN, et al. Development of novel alkene oxindole derivatives as orally efficacious AMP-activated protein kinase activators. ACS Med Chem Lett 2013; 4: 475–80.
Rajamohan F, Harris MS, Frisbie RK, Hoth LR, Geoghegan KF, Valentine JJ, et al. Escherichia coli expression, purification and characterization of functional full-length recombinant alpha2beta2gamma3 heterotrimeric complex of human AMP-activated protein kinase. Protein Expr Purif 2010; 73: 189–97.
Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol 2009; 297: C1041–52.
Liu J, Chen D, Liu P, He M, Li J, Li J, et al. Discovery, synthesis, and structure-activity relationships of 20(S)-protopanaxadiol (PPD) derivatives as a novel class of AMPKalpha2beta1gamma1 activators. Eur J Med Chem 2014; 79: 340–9.
Luo C, Long J, Liu J. An improved spectrophotometric method for a more specific and accurate assay of mitochondrial complex III activity. Clin Chim Acta 2008; 395: 38–41.
Qiu BY, Turner N, Li YY, Gu M, Huang MW, Wu F, et al. High-throughput assay for modulators of mitochondrial membrane potential identifies a novel compound with beneficial effects on db/db mice. Diabetes 2010; 59: 256–65.
Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, et al. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 2006; 574: 41–53.
Kim M, Hunter RW, Garcia-Menendez L, Gong G, Yang YY, Kolwicz SC Jr, et al. Mutation in the gamma2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res 2014; 114: 966–75.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 81502910, 81273566 and 81673489), the Shanghai Commission of Science and Technology (No 15ZR1447900 and 14431902800), the Youth Innovation Association of Chinese Academy of Sciences (Mei ZHANG), and the National Key Research and Development Program of China (No 2016YFC1305500).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Supplementary Materials and methods
Supplementary Figures and Table
Rights and permissions
About this article

Cite this article
Zhang, M., Xie, Zf., Zhang, Rt. et al. Novel substituted pyrazolone derivatives as AMP-activated protein kinase activators to inhibit lipid synthesis and reduce lipid accumulation in ob/ob mice. Acta Pharmacol Sin 39, 1622–1632 (2018). https://doi.org/10.1038/aps.2017.186
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.186
Keywords
This article is cited by
-
TLR2 Mediates Microglial Activation and Contributes to Central Sensitization in a Recurrent Nitroglycerin-induced Chronic Migraine Model
Molecular Neurobiology (2023)
-
Pyrazolone derivative C29 protects against HFD-induced obesity in mice via activation of AMPK in adipose tissue
Acta Pharmacologica Sinica (2021)
